重榜盘点!中国引用量最高的10篇生命科学文章系统解析
这次系统地解析了10篇中国生命科学最高引用的文章。
我们发现华大基因王俊等团队2篇,中科院北京基因组研究所杨焕明等团队(原单位)2篇;病毒基因工程国家重点实验室侯云德等团队贡献了2篇;国家癌症控制中心赫捷等研究组1篇;中日友好医院何江等团队1篇;南京大学张辰宇1篇;同济大学医学院1篇。这10篇的主要内容包括5篇基因组序列的组装,2篇流行病学(糖尿病及癌症),1篇技术的开发,1篇miRNA潜在的医学应用,1篇抗癌药物的临床评判。
现在我们按引用量进行排序,进行解读,以飨读者。
1.通过宏基因组测序建立人类肠道微生物基因目录(时间2010年,引用次数3502)


要了解肠道微生物对人类健康和福祉的影响,评估其遗传潜力至关重要。在这里,华大基因王俊等团队描述了基于Illumina的宏基因组测序,330万个非冗余微生物基因的组装和表征,其中是来自124个欧洲个体的粪便样品,总共得到576.7kMb的序列。
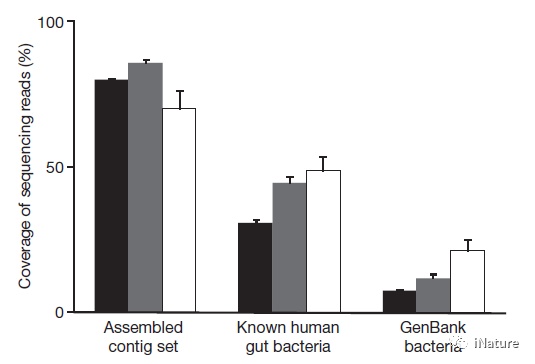
该基因集合与人类基因相互补,其中含有绝大多数的流行(更频繁)的人群微生物基因,并可能包括大部分流行的人类肠道微生物基因。这些基因大部分是在队列中分享的。超过99%的基因是细菌,表明整个人群中存在1,000到1,150种流行细菌物种,每个个体至少有160种这样的物种,这也是基本共享的。
2.籼稻和粳稻基因组的再次组装(时间2005年,引用次数2918)

杨焕明等研究组报告了籼稻和粳稻基因组的鸟枪法全基因组序列,比2002年的草案提高了近1000倍。测试了19,079个全长cDNA的非冗余集合,97.7%基因在没有碎片的情况下与一个或另一个基因组的能够被定位到。杨焕明等研究组引入了一种基因鉴定程序,不依赖于已知基因的相似性来消除由转座因子引起的错误预测。使用可用的EST数据来调整预测中的残差,估计的基因数至少为38,000 - 40,000。只有2%-3%的基因对于任何一个亚种都是独特的,与可能仍然缺失的序列量相当。
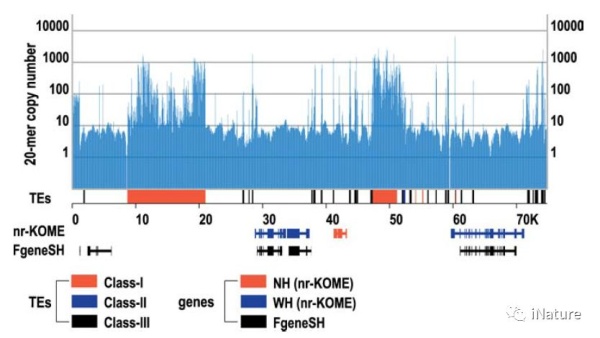
尽管基因含量缺乏变异,但是基因间区域有很大的变异。杨焕明等研究组发现了18个不同的重复片段,覆盖了65.7%的基因组;这些对中的17个可以追溯到草发散之前的一个普通时间。更重要的是,正在进行的个体基因复制为基因发生提供了永无止境的原料来源,并且是草族成员之间差异的主要贡献者。
3.痢疾志贺氏菌亚群的比较基因组学研究与进化分分析(时间2005年,引用次数2752)


志贺氏菌引起细菌性痢疾,这仍然是公共卫生的重大威胁。属性状态和物种分类不再有效。尽管如此,1型痢疾杆菌致病致死,而志贺氏菌仅限于印度次大陆,福氏志贺氏菌和宋内志贺菌分别在发展中国家和发达国家流行。为了在基因组水平开始解释这些独特的流行病学和病理学特征,病毒基因工程国家重点实验室侯云德等团队对4个代表性菌株进行了比较基因组学研究。每个志贺氏菌基因组包括编码保守的主要毒力决定簇的毒力质粒。
志贺氏菌染色体与大肠杆菌K12菌株MG1655的基因大部分共享,但每个基因组都有200多个假基因,300个与700个拷贝的插入序列(IS)元件相似,以及大量的缺失,插入,易位和倒位。假定的毒力基因具有广泛的多样性,主要是通过噬菌体介导的横向基因转移获得的。因此,通过包括噬菌体介导的基因获取,IS介导的DNA重排和假基因形成的收敛性进化涉及功能的获得和丧失,志贺氏菌属成为具有不同流行病学和病理特征的高度特异性人类病原体
4.中国男性和女性糖尿病患病情况(时间2010年,引用次数2649)


由于中国生活方式的急剧变化,人们担心糖尿病可能会流行起来。中日友好医院何江团队从2007年6月至2008年5月进行了一项全国性研究,以评估中国成年人中糖尿病的流行情况。总糖尿病(包括先前诊断的糖尿病和既往未确诊的糖尿病)和糖尿病前期的年龄标准化患病率分别为9.7%(男性为10.6%,女性为8.8%;男性为5020万,女性为4220万)和15.5%(男性为16.1%,女性为14.9% ;男性为7610万,女性为7210万)。
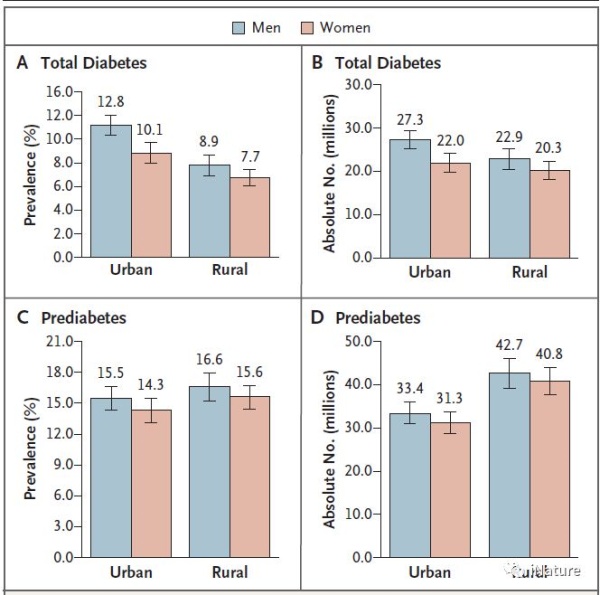
糖尿病患病率随着年龄的增长而增加(分别为20〜39岁,40〜59岁和> 60岁者分别为3.2%,11.5%和20.4%),随着体重的增加而增加。城市居民糖尿病患病率高于农村居民(11.4%比8.2%)。单纯性糖耐量异常的患病率高于单纯性空腹血糖异常(男性为11.0%比3.2%,女性为10.9%比2.2%)。这些结果表明,糖尿病已成为中国主要的公共卫生问题,需要针对糖尿病的防治策略。
5.癌症在中国的流行病学(时间2016年,引用次数2449)

随着发病率和死亡率的增加,癌症是中国的主要死亡原因,是一个重大的公共卫生问题。由于中国人口众多(13.7亿),以前的国家发病率和死亡率估计数据仅限于使用1990年代数据或特定年份数据的小样本人群。通过中国国家中央癌症登记处现有的更多人口登记数据库提供的高质量数据,国家癌症控制中心赫捷等研究组分析了来自72个当地以人口为基础的癌症登记处(2009-2011)的数据,占人口总数的6.5%,用来估计2015年新发病例和癌症死亡人数.22个登记处的数据用于趋势分析(2000-2011年)。结果显示,2015年中国将有429.2万新增癌症病例和2814000例癌症死亡,其中肺癌是最常见的癌症,也是导致癌症死亡的主要原因。
胃癌,食管癌和肝癌也被普遍诊断并被确定为癌症死亡的主要原因。农村居民的年龄标化(Segi人群)发病率和死亡率显着高于城市居民(分别为213.6 / 10万和191.5 / 10万,发病率分别为149.0 / 10万和109.5 / 10万)。对于所有合并的癌症,男性在2000年至2011年期间发病率稳定(每年增加0.2%; P = 0.1),而女性则显著增加(每年增加2.2%; P<0.05)。相比之下,2006年以来的死亡率在男性(每年-1.4%,P <0.05)和女性(每年-1.1%,P <0.05)均显著下降。许多估计的癌症病例和死亡可以通过减少危险因素的流行来预防,同时提高临床护理服务的效率,特别是对于生活在农村和处于不利地位的人群。
6.血清中miRNA的特征:用于诊断癌症和其他疾病的新型生物标志物(时间2008年,引用次数2443)

miRNA在各种组织中的失调表达与多种疾病(包括癌症)有关。在这里,张辰宇等研究组证明miRNA存在于人类和其他动物(如小鼠,大鼠,牛胎儿,小牛和马)的血清和血浆中。血清中miRNA的水平是稳定的,可重复的,并且在同一物种的个体中是一致的。使用Solexa,张辰宇等研究组对健康中国受试者的所有血清miRNA进行测序,发现男性和女性受试者中分别有超过100和91个血清miRNA。
张辰宇等研究组还鉴定了肺癌,结肠直肠癌和糖尿病的血清miRNA的特定表达模式,提供了血清miRNA含有各种疾病指纹的证据。通过Solexa获得的两个非小细胞肺癌特异性血清miRNA在75名健康供体和152名癌症患者的独立试验中进一步验证。通过这些分析,张辰宇等研究组得出结论:血清miRNA可以作为检测各种癌症和其他疾病的潜在生物标志物。
7.水稻基因组草图(时间2002年,引用次数2230)


杨焕明等团队通过全基因组鸟枪测序为中国栽培最广泛的亚种 - 水稻(Oryza sativa L. ssp。)制定了水稻基因组序列草图。 基因组大小为466Mb,估计有46,022至55,615个基因。 装配序列的功能覆盖率为92.0%。 大约42.2%的基因组是在精确的20个核苷酸的寡聚体重复中,并且大多数转座子位于基因之间的基因间区域。 尽管预测的拟南芥基因中有80.6%在水稻中具有同源物,但预测的水稻基因中仅有49.4%在拟南芥中具有同源物。
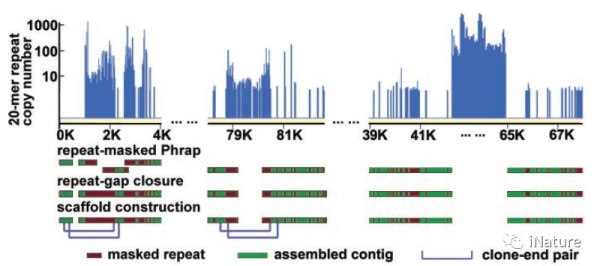
8.福氏志贺氏菌血清型2a的基因组序列(时间2002年,引用次数2023)


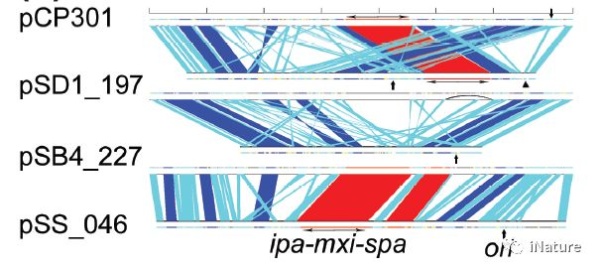
病毒基因工程国家重点实验室侯云德等团队对福氏志贺氏菌血清型2a的基因组进行了测序,福氏志贺氏菌血清型2a是人类中导致细菌性痢疾或志贺菌病的最普遍的物种和血清型。全基因组由4607203 bp的染色体和221618 bp的毒力质粒组成,命名为pCP301。虽然该质粒显示出与血清型5a测序的较小偏差,但已经揭示了染色体的显著特征。

S.flexneri染色体惊人地具有314个IS元件,是其近亲,非致病K12菌株和肠出血性O157:H7大肠杆菌菌株的7倍以上。与E.coli序列相比,共有13个易位和倒位,均含有大于5kb的片段,大部分与缺失或获得的DNA序列有关,其中有几个可能是噬菌体传播的致病岛。此外,S.flexneri类似于另一种人类受限的肠道病原体伤寒沙门氏菌,与大肠杆菌菌株相比也具有数百个假基因。所有这些都可能受到进一步观察,以针对志贺菌病的新型预防和治疗策略。
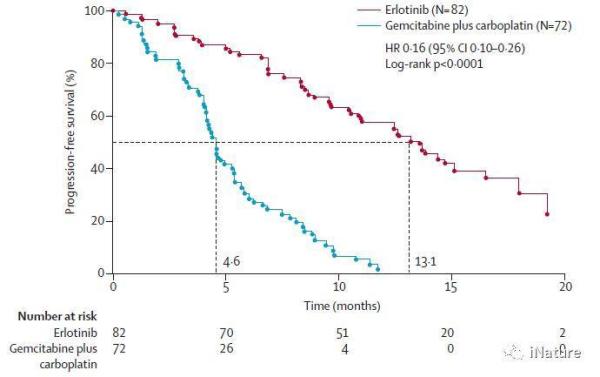
9.埃罗替尼与化疗作为EGFR突变阳性非小细胞肺癌患者的一线治疗的临床试验(时间2011年,引用次数1876)


激活EGFR突变是非小细胞肺癌(NSCLC)中酪氨酸激酶抑制剂(TKI)治疗反应的重要标志物。 OPTIMAL研究比较了TKI厄洛替尼与标准化疗在晚期EGFR突变阳性NSCLC患者的一线治疗中的疗效和耐受性。同济大学医学院等团队揭示与标准化疗相比,厄洛替尼使晚期EGFR突变阳性的非小细胞肺癌患者获得显著的生存时间延长,并且具有更好的耐受性。 这些发现提示厄洛替尼对晚期EGFR突变阳性的非小细胞肺癌患者的一线治疗非常重要。
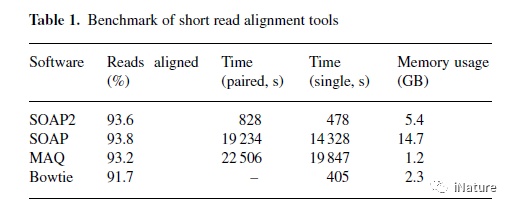
10.SOAP2:用于短读取对齐的改进的超快工具(时间2009年,引用次数1759)


SOAP2是短寡核苷酸比对程序的显著改进版本,其既减少计算机内存使用量又以前所未有的速率增加对准速度。 王俊等研究组使用Burrows Wheeler Transformation(BWT)压缩指数替代种子策略来索引主存储器中的参考序列。 王俊等研究组对整个人类基因组进行了测试,发现这种新算法将内存使用量从14.7降低到5.4GB,并将对齐速度提高了20-30倍。 SOAP2兼容单端和双端读取。 此外,这个工具现在支持多个文本和压缩格式。 共识建立者也已经被开发出来用于在参考基因组上进行短读数比对的共有装配和SNP检测。

来源:iNature
版权及免责声明:本网站所有文章除标明原创外,均来自网络。登载本文的目的为传播行业信息,内容仅供参考,如有侵权请联系答魔删除。文章版权归原作者及原出处所有。本网拥有对此声明的最终解释权。
{replyUser1} 回复 {replyUser2}:{content}