Car-T细胞治疗产品生产的挑战和考量
尽管近两年,Car-T细胞治疗技术得到了飞跃式的发展,其具有巨大的潜力,未来可能可用于针对肿瘤的标准医疗体系。但从许多方面来看,获得性T细胞免疫仍处在起步阶段,T细胞生产的工艺相当复杂,其由多个步骤组成,包括T细胞富集、活化、转导、扩增以及制剂/冻干。每个步骤又受多种因素的影响,包括“种子”或起始细胞群落的组成、T细胞活化的方法、抗原受体转基因递送载体、基因编辑的效率、细胞处理能力以及最终的产品控制。厂商、科研以及监管等团队之间需要持续的合作,聚焦于一个可获得持续改进的“反馈”回路,驱动工艺的优化,最终获得可“真正”商品化的Car-T细胞治疗产品。
Car-T细胞治疗产品生产的挑战和考量(J.Xu, et al, 2018)
T细胞生产的可重复性以及可放大性
最开始的大部分的Car-T细胞生产设施位于科研机构中,如美国的宾州大学以及国家癌症研究所。许多这样的学术中心都会选择跟不同规模的制药公司形成战略性的合作伙伴关系,目标是细胞治疗产品的商业化。其生产设施旨在获得患者特异性的细胞产品,用于I期和II期临床试验,但这种生产过程依赖于熟练的细胞生产人员以及数个开放式操作步骤。在这些开放式操作步骤中,即使极小程度的变异性,也会影响最终细胞产品的质量。所以,复制高度依赖人工的工艺程序,对于Car-T细胞生产的规模扩展仍是一个主要的挑战。而又因为Car-T细胞可用于靶向不同类型的肿瘤,转基因递送载体和最终细胞产品的生产规模还取决与每种适应症的发生率。此外,对于可预测的细胞基因工程,一致、稳健的转基因递送平台的生产必需考虑基因治疗的长期安全性以及预期的法规限制。
提高细胞生产效率
围绕Car-T细胞治疗的知识的爆发性增长,使得对如何提高每个Car-T生产步骤效率有了更深入的了解。此外,临床T细胞生产时,还必需考虑以下基本要求。首先,生产过程必需优化,以获得可用于患者的安全且稳健的细胞产品。来自患者的功能性T细胞的优化筛选以及扩增/培养(包括T-细胞刺激因子、培养基、细胞因子的使用以及培养系统中药理调节剂的添加)仍是一个不小的挑战。其次,优化生产过程的稳健性,降低人工工作量,同时使污染的风险最小化,提高处理能力,促进T-细胞生产的标准化。

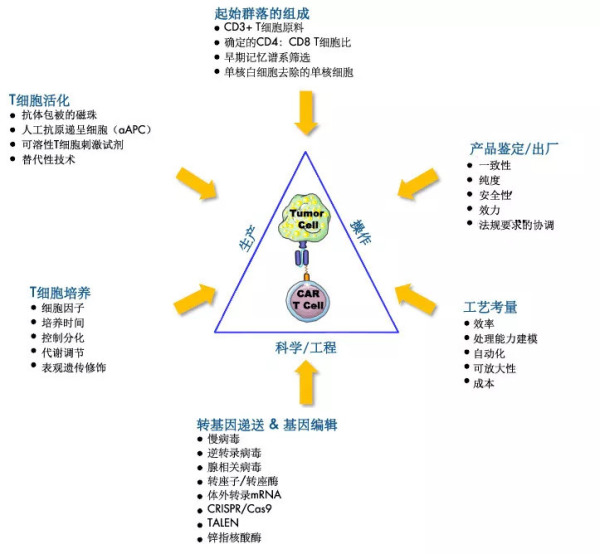
T-细胞生产的挑战和可能的解决方案(J.Xu, et al, 2018)
自动化的细胞生产
考虑到自体T细胞生产需要耗费相当的人力,且每个步骤需要多个仪器,加上昂贵的试剂以及多个开放式操作步骤,降低可能影响可重复性、效力以及安全性的变异性至关重要。从这方面考虑,最近开发的自动化系统可能是非常有潜力的解决方案。多种半自动化或全自动化的设备被开发用于不同的工艺步骤,包括用于细胞分离、细胞漂洗以及浓缩的设备。自动化同时可提供在每个生产步骤进行过程中采样以及质量控制的机会,而且,自动化可允许在生产过程中,方便地进行检测和文件记录。在不久的将来,更多自动化生产设备以及整合这些设备的新工艺的出现,可能可提供更多标准化的细胞产品,以支持Car-T细胞治疗的临床应用。
用于T细胞生产的基因编辑技术
另一种简化T-细胞治疗的方法是生成普适性的通用“现货型”T细胞产品,用作获得性细胞治疗,从而服务更大的患者群体。新的基因编辑技术的出现,包括锌指核酸酶(ZFN)、转录激活子样效应核酸酶(TALEN)以及最近出现的CRISPR/Cas9,已经为生产异源性通用“现货”商品化规模细胞治疗铺平了道路。使用“现货”通用型T-细胞产品,我们可以克服患者来源的细胞由于较差的质量、产量和/或疾病状态,而不能生成T-细胞产品的问题。这将降低生产成本和时间,允许获得可重复的、有效力的T-细胞产品。更重要的是,通过靶向内源性αβ TCR和MHC I类分子HLA,可降低αβ T细胞介导的移植物抗宿主疾病(GVHD)的风险。除了异源性T细胞,通过基因编辑技术,干扰内源性TCR也有利于生产自体TCR-T细胞产品,以降低内源性和重组TCR之间的TCR错配。如基于CRISPR/Cas9和相似的基因编辑技术在大规模T细胞生产中的使用和优化,预期在不就的将来,可见到普适性T-细胞产品在临床中使用。
与细胞生产相关的成本
用于人体治疗性应用的细胞产品必需在符合监管机构指南的GMP条件下生产。此类细胞产品的所要求进行的大量测试产生的成本通常相当惊人,是其商品化的另一个挑战。除了一致性、纯度和效力评估,通过整合病毒而进行修饰的细胞产品,按FDA指南,如果培养液在转导后96小时内收获,还必需筛选具有复制能力的逆转录病毒和慢病毒载体(RCR/RCL)。基于细胞培养的方法是目前用于输液产品中RCR/RCL检测的标准方法,操作时,待测样品与特定细胞系(如用于慢病毒的C8166细胞)共培养至少3周或以上,以进行病毒扩增,随后进行RCR/RCL的终点检测。但是,这些严格的共培养分析耗时耗力,成本也很高。此外,基于q-PCR的病毒成分或转基因的检测是另一种快速且经济的替代方法,可对RCR/RCL进行初步筛选。qPCR和生物学RCR/RCL检测均已广泛用于筛选临床病毒载体批次和生产的T细胞产品。但是截至目前,在使用第三代慢病毒载体和逆转录病毒载体的临床试验中,还没有阳性RCR/RCL结果的报导,说明输注产品中RCR/RCL的形成可能性不高。所以,从另一方面来讲,可能需要重新评估针对RCR/RCL的指南。
细胞生产的另一个主要成本是人血清,其为T细胞存活和生长提供基本要素。商品化的人血清价格昂贵,且供应量有限。此外,必需对批次间的差异进行评估,以符合法规和效力要求,且输注前细胞产品中存在的血清可能引起患者的不良反应。所以,非动物源性的人血清替代物在细胞生长背景下可能是必要的。目前已有此类商品化产品,预期出于经济性目的,其将整合至当前的T-细胞生产,以维持或提高T-细胞产品质量。
预期的法规指南
尽管监管机构,如FDA,多年来一直在规范细胞和基因治疗,并形成了针对产品检测和出厂标准等的指南文件,但不同国家和地区的这些法规和文件可能并不完全一致,用于临床应用的基因修饰细胞产品的检测和出厂成本可能会因为特定监管机构的关注点不同而有很大的差异。所以,当细胞治疗产品旨在服务全球化的患者群体时,从全球化的水平,来协调法规要求,将在基于细胞/基因治疗的监管中标准化最佳实践作为目标,将极大地有助于形成安全、高效且经济的平台。

来源:生物制品圈
版权及免责声明:本网站所有文章除标明原创外,均来自网络。登载本文的目的为传播行业信息,内容仅供参考,如有侵权请联系答魔删除。文章版权归原作者及原出处所有。本网拥有对此声明的最终解释权。
{replyUser1} 回复 {replyUser2}:{content}