阻碍CAR-T细胞治疗若干因素
“2012年4月六岁的Emily Whitehead,接受了CAR-T-CD19注射治疗ALL,获得持续缓解,至今持续生存。2018年诺华CAR-T细胞疗法Kymriah和Kite Yescarta先后获批。但是CAR-T治疗缓解患者一年后,30-50%复发,且还有10-20%的患者对于CAR-T治疗不敏感,因而需要系统归纳细胞治疗耐药的因素。”
CAR-T 细胞生产
一个产品生产的障碍来自于病人T细胞收集。因为病人在接受CAR-T治疗之前,多数接受过化疗等细胞毒性治疗,导致T细胞数量下降,现在还没有数据,到底多少比例的病人,其T细胞数量够T细胞治疗。
通常进入PhaseI的儿童患者,绝对淋巴细胞计数中位数是1228细胞/μl,美国国家癌症研究所,建议CD3+T细胞数量大于150细胞/μl,即可进行CAR-T细胞生产。一项56例的CD22临床治疗,55例病人的CD3+T细胞中位数567 cells/μl (范围145–2,144 cells/μl),绝对淋巴细胞计数775 cells/μl (范围 230–4,620cells/μl)无疑获取到足够的T细胞是治疗的前提。
化疗药物除了影响病人T细胞数量,也会影响获取的T细胞的质量,如接受过阿霉素,氯法拉滨。而环磷酰胺和阿糖胞苷治疗,可以减少早期发育的T细胞(early lineage T cells),这群细胞与CAR-T细胞的扩增密切相关。
实体瘤病人,比血液肿瘤收取T细胞的难度大,因为在循环的细胞抑制性T细胞的比例更高。对于有高复发预期的病人,或者复发病人在治疗前,进行T细胞收集有助提高CAR-T生产的成功率。
现在CAR-T生产的流程中,大多需要通过TCR进行细胞活化,在FDA批准的CAR-T-CD19产品中,都使用anti-CD3,anti-CD28抗体来活化扩增T细胞。在临床阶段的大部分产品也在用这一方法进行T细胞活化扩增。除此之外,还包括一些细胞因子活化的方法在用,比如加入IL-2,IL-7,IL-15等。但是哪一种方法更好,没有定论。
现多数CAR-T细胞产品是CD4+,CD8+T 细胞的混合物,且可能不同病人之间两种细胞的比例差别非常大,虽没有数据显示比例会影响疗效,但是对于细胞产品的质控带来不便。已经开始的一些临床研究,开始使用CD4+ ,CD8+分离的产品,按照固定比例输给病人。现在全自动的封闭的生产系统使用对于提高生产,也是一个趋势。
此外,即使CAR-T细胞被生产出来,其其实的T细胞表型,对于后续的临床疗效也是至关重要
比如中枢记忆,及干细胞样记忆T细胞,因而生产工艺的控制产生特殊表型的CAR-T,也是产品成功的重要因素。一项接受CD-19CAR-T 治疗的CLL研究中,应答者和无应答者,区别在于记忆基因更丰富,有更高比例的高表达IL-6R的CD27+PD-1?CD8+ CAR T,这会导致更好的肿瘤控制,因而这一表型是治疗中更期待的。
分离细胞的异质性也是一个挑战,比如高比例的来自于髓系的抑制性细胞的存在,他们可以抑制T细胞的生长。因而开始有一些小组通过更好的磁珠分选手段,获取均一性更好的T细胞初始样品。
CAR结构设计是另一个可能影响CAR T细胞产品表征和修饰细胞体内行为的因素,包括其扩张动力学和持续时间。在临床试验中测试的大部分CAR t细胞产品都是第二代药物,它们既含有TCR刺激域(通常来源于T细胞表面糖蛋白CD3ζ-ζ),也包含单个共刺激结构域。目前FDA批准的产品含有CD 28或4-1BB(也称为CD 137)共刺激域。共刺激域对应答率的影响还没有得到系统的评价,尽管临床前的数据和患者的观察表明,CAR设计的这一方面显着地影响了细胞产物的持久性。CAR设计的其他细节,如抗原结合区的特定特征,细胞外铰链区的存在和结构,以及跨膜区的特征,也可能影响CAR-T细胞的属性,但尚未产生明确的数据来定义这些设计细节的影响。
CAR基因载体转染载体的选择。现在FDA批准的主要使用逆转录病毒载体,此外还有HIV衍生的慢病毒载体。载体的选择涉及转染效率,CAR-T细胞活力等。RNA瞬时转染也在临床前被使用,但不用于临床产品。此外,转座子,转座系统也被用于临床级别的产品,也被证实了其有效性和安全性。
CAR-T细胞效能评价,尤其是特定标志物的寻找,在这方面的进展还是比较缓慢。鉴于白血病患者对CART细胞治疗的高反应率,数量较少的无反应者,系统地评估和确定与缺乏反应性有关的参数是困难的-特别是考虑到根本原因可能是多因素的,而不完全只能归因于产品变量。然而,在CLL患者中,其应答率远低于ALL或淋巴瘤患者,Fraietta等人确定有利的产品特征,如在CAR-T细胞生成之前,IL-6-STAT 3信号的富集和CD29+CD45RO?CD8+T细胞的频率升高。此外,输注前的产品,细胞因子和趋化因子表达谱的由多功能T细胞亚群组成,与无多功能T细胞亚群的产品相比,在淋巴瘤患者中的反应有所改善。为了建立CAR-T细胞的理想属性,需要更多的数据,但最优特性可能因car结构和目标的恶性程度不同而有所不同。
从健康供体采集T细胞,进行异体移植,可能是另外一个选择。已经有多个临床研究开始。这个策略,通常需要使用基因编辑的方法降低移植排斥,但是因为提高了T细胞质量,国内外都有多家企业和研究机构在进行,包括CAR-T,CAR-NK等产品。
CAR-T 细胞输注
如果患者疾病进展前或者疾病有关的并发症出现前,不能及时注入CAT-T细胞,可能妨碍成功的治疗。审查了FDA批准的,CD19CAR-T细胞产品,在儿童和ALL年轻病人中的试验,92名患者中,有17名(18%)没有接受CART细胞注射,原因包括7名患者(主要是由于细胞生长不良)出现与tisagenlecleucel有关的产品问题,7名患者死亡(4名患者因疾病进展和其他患者感染相关并发症而死亡),以及导致患者没有资格接受CAR-T细胞注射的不良事件(真菌病或GVHD)。在本研究中,细胞输注的中位时间为45天(范围:30-105天)。
同样,在纪念斯隆·凯特林癌症中心进行的一项研究中,在78名ALL患者中,11名(14%)没有进行细胞制造(大多数人寻求替代治疗),在67名接受CART细胞制造的患者中,有13名(19%)没有接受输液,部分原因是产品失败、疾病进展或并发症使他们没有资格接受治疗。由于20%-30%的候选细胞最终没有被输注CAR-T细胞,未来需要寻找缩短生产时间的策略,以提高符合条件的患者接受输液的可能性,从而增加受益于CART细胞治疗的患者人数。
CAR-T 细胞活化及扩增
临床研究表明,有效治疗所需的CAR t细胞的剂量非常小,目前的给药方案为0.2~5.0×106个/kg或每次输注0.1-2.5×108个转导的CART细胞,但输注后细胞的活化和指数扩张是必需的,质量和固有的T细胞表型CAR-T细胞产品可能会影响输注后的CAR-T细胞行为。
此外,接受治疗的病人体制相关因素也是CART细胞扩张的重要因素.例如,疾病(以及抗原)负担会对细胞的增殖程度产生积极的影响,这反过来又可能增加CRS的风险和严重性。淋巴耗竭对CAR-T细胞的扩张也很重要,有证据表明,某些化疗药物,例如氟达拉滨,在这方面可能更有效。然而,氟达拉滨被认为是CAR-T细胞相关神经毒性的潜在贡献者之一,尽管经典的氟达拉滨相关神经毒性的发生时间与CAR-T细胞相关毒性不同,而且这种药物对淋巴耗竭的安全性普遍支持。
接受CAR-T 细胞治疗的门槛
尽管FDA在过去两年中批准了CD 19靶向的CART细胞产品,但获得这些新疗法的机会仍然有限。随着FDA批准了两种不同的抗CD 19 CAR-T产品,有资格进行CAR-T治疗中心越来越多,目前在美国有超过73个单独的中心。因此,符合条件的患者在当地癌症中心接受FDA批准的CAR-T细胞产品的能力正在提高,从而促进将这些疗法纳入他们的个人治疗计划。
此外,费用和保险范围是扩大患者获得CART细胞治疗的持续障碍。成本分析和优化生产策略,以降低成本是必要的。
许多患者将继续寻求参与临床试验,以避免在获得FDA批准后适应症的限制。
靶抗原阳性的复发
早期所有复发,通常是在成功诱导缓解后的头几个月内,与有限的CAR-T细胞持久性和/或短暂的B细胞发育不全有关,这意味CART细胞介导的对白血病的监视活性的丧失。CAR-T细胞持久性的决定因素仍有待完全确定,除了固有的T细胞质量和初始T细胞表型(包括CD4与CD8 T细胞的比例)外,还包括构建在CAR结构中的共刺激域,而临床前的报告显示,含有CD28共刺激域的CAR持久性不如包含4-1BB共刺激域的CAR;临床经验与这些数据是一致的。临床数据显示,含4-1BB的CAR-T在体内持续时间中位数是168天(范围:20-617天),而CD28只有30天,且3个月后就很少能检测到了。4-1BB的信号可能会降低T细胞衰减的速度。其他能够获取更好CAR-T持久性的共刺激分子也是研究的重要方向。
CRISPR–Cas9技术的使用,可以将CAR特异性加入到TCRa恒定区域,可以获得更好的效果,也是下一代CAR设计的重要手段。
提高CAR-T持久性的设计和生产在临床上开始使用,比如注射T细胞-抗原递呈细胞(T-APCs),在病人缓解后,定期刺激活化CAR-T,以确定反复刺激是否能重新激活和在数量上扩展CART细胞,防止抗原阳性复发(NCT03186118)。更广泛地说,人工抗原提呈细胞的使用,为优化过继性T细胞免疫治疗提供了一种潜在的方法,提高了注入的T细胞的治疗效果和持久性。促使CAR-T细胞向中央记忆或干细胞样记忆表型迁移,是增强治疗反应和细胞持久性的另一种独特方法。
CAR-T和免疫检查点抑制剂及其他免疫调节方法一起使用,为优化临床应答的发生率、深度和耐用性提供了一种协同方法。临床标本表明,PD-1在CAR T细胞中的表达增加,临床数据支持PD-1-PD-L1阻断在提高CAR T细胞治疗效果中的作用,证明了这种方法对持久性改善。
CD19 CART细胞持久性对ALL患者的持久缓解很重要;然而,是否对所有CART细胞产品所实现的持久缓解的都很重要,目前还不清楚。临床证据表明CART细胞持久性的相关性可能因癌症类型而异。
抗原阳性病人复发,采用再输注CAR-T细胞的策略,因为失去了CAR-T的持久性,成功比例降低。Gardner et al等的一项研究,10个ALL成人及儿童接受anti-CD19 CAR-T再输注,8个失去了持久性,2例出现CAR-T扩张,仅1例出现缓解。后续又有多个类似低或者无反应性的临床报道。有报道,在CAR-T受试者中,出现抗CAR-T的T细胞,显示免疫排斥可能是一个重要原因。因而采用环磷酰胺外,及含氟达拉滨的淋巴耗竭方案可以改善再灌注反应,以及改善最初的CAR-Tt细胞扩张和持久性。也有开始使用全人源CAR,或者换另外一个靶点的CAR等方式来改善。
靶抗原丢失或者突变引起的免疫逃逸
治疗缓解病人,肿瘤发生靶抗原丢失或者表达量降低,都会引起相应的免疫逃逸,从而产生患者复发。在ALL病人已经有很多描述了,在脑胶质瘤等实体瘤也有类似的描述。在未来的研究和新的CAR结构的设计中,需要认识抗原密度在抗肿瘤反应中的作用,并确定导致靶表达中断的机制,以优化CAR T细胞的反应。
其他靶向免疫治疗可能进一步增加CART细胞治疗后免疫逃避。例如,抗CD 19双特异性抗体blinatumomab和抗CD 22 ADC药物inotuzumab,都是FDA批准的治疗ALL的疗法,均由于使用后出现CD 19-或CD 22-而失去反应。因此,这些药物可能会降低,针对相同抗原的CAR T细胞的治疗效果,或者降低反应的持久性。所以接受博纳吐抗体药物治疗,是ALL病人接受CAR-T治疗的排除因素。
除了治疗靶向抗原的丢失之外,肿瘤本身异质性非常强。比如CD19被认为是所有Pre-B 细胞表达的抗原,但是随着研究的深入发现了一些罕见的CD19-的Pre-B细胞克隆,尤其是在BCR–ABL1 ALL,这时anti-CD19就无法对于此群Pre-B细胞产生治疗效果。CD22也有类似的问题。
此外细胞谱系变换,也是另外一个引起肿瘤逃逸的机制。有报道,ALL病人特异性治疗后,因为KMT2A重排,会变为AML。此外在临床前ALL模型,靶向FLT3的CAR-T治疗后,发现B细胞系向T细胞系的转换。在非淋巴系列肿瘤中,是否存在这种机制,还有待继续研究。
尽管抗原调节作为一种免疫逃逸机制得到了重视,但目前无法预测哪些患者有较高的发生抗原调节,产生复发疾病的风险,除了那些已知的机制:确认存在的抗原阴性亚克隆,那些在抗原表达上已经存在异质性的患者,或者那些曾经接受过针对同一抗原的免疫治疗的患者。更精准的针对细胞表型的流式细胞术的开发,有助于解决一部分问题。
鉴于抗原调节作为逃避有效免疫治疗的机制的倾向,结合多抗原靶向的CAR结构正在被开发,以解决固有的肿瘤异质性,从而降低白血病复发的风险。多靶点方法的临床前数据,包括使用串联抗CD 19-CD 20 CAR结构、组合抗CD 19和抗CD 123(又称il-3rα)策略,CD 19和CD 22,使用针对两种抗原的单个CAR结构或针对每种抗原的两种独特的CAR,这些策略的一些临床试验正在进行中。这些方案的后续数据将有助于确定双抗原靶向治疗方法是否足以防止疾病复发,或者是否需要针对两种以上抗原的额外组合策略,以使CART细胞治疗有效。在组合多抗原靶向策略的开发中,确保对每种抗原的有效反应是至关重要的:优先选择一种抗原而不是另一种抗原可能导致对单一抗原的功能反应的偏好,而不排除抗原阴性复发的问题。然而,开发功能性多目标结构并非易事,它高度依赖于临床前测试,以确定具有同等能力同时靶向抗原的生物活性结构。
CRS引起的病理变化
在肿瘤细胞被CAR T细胞识别后,T细胞的激活和增殖伴随着活化的淋巴细胞和其他免疫细胞(包括单核/巨噬细胞、树突状细胞、可能的基质细胞和内皮细胞)释放细胞因子。典型情况下,CRS发生在CAR-T细胞治疗后的第2~14天,随后的时间足以使CAR-T细胞扩张、循环到肿瘤部位并执行其最初的效应功能,包括细胞因子的产生。CRS很少发生在CAR-T细胞输注后2周以上,通常在CRS发病后2~3周内消退。CRS具有高水平的血清细胞因子和炎症标志物,特别是IL6、IFN-γ、铁蛋白和C反应蛋白的特点。临床上,症状包括高烧和流感样症状,可发展为低血压,毛细血管漏,缺氧和多器官功能障碍。
其他细胞因子可能在CRS中升高,包括TNFα、IL 10、IL 2Rα等。虽然IL-6被认为是CRS的重要介质,但其发病机制的细节仍不清楚。最近的数据表明,CAR-T细胞不是IL-6的主要来源,而是由激活的髓样细胞、内皮细胞和基质细胞分泌的。此外,小鼠模型提示IL-6对于CAR-T细胞功能是可有可无的,从而引起了对该功能的关注在CRS管理中通过IL-6阻断减轻疗效的理论风险。
细胞因子释放综合征与神经毒性相关。临床经验和改善的病理生理模型都表明,严重的细胞因子释放可能会增加神经毒性的风险。回顾弗雷德·哈钦森癌症研究中心(FHCRC)接受CART 19治疗的133名成年人的数据,发现严重神经毒性患者有内皮激活的证据,包括弥散性血管内凝血(DIC)、毛细血管渗漏和血脑屏障(BBB)通透性增高。在神经毒性患者中,配对血清和脑脊液标本显示TNF-α、IL-6和IFN-γ水平升高,提示血脑屏障允许高浓度的全身细胞因子进入脑脊液,或这些细胞因子是通过浸润T细胞局部产生的。细胞因子在脑脊液中的存在可能产生一种前馈状态,肿瘤坏死因子α和干扰素γ诱导脑血管病周细胞应激和周细胞分泌内皮细胞激活的细胞因子,包括IL-6和血管内皮生长因子,从而进一步增加血脑屏障通透性。
对一位具有致命神经毒性的病人的病理评估发现,大脑中内皮激活和多灶性血管破裂,类似于非人类灵长类动物的发现。≥4级神经毒性患者治疗前早期血清细胞因子水平或内皮活性的生物标志物均有较高的接受率。这些发现提供了一个洞见,严重的CRS可能随后导致神经毒性的发展,并加强了对CRS的密切警惕和早期治疗的必要性。
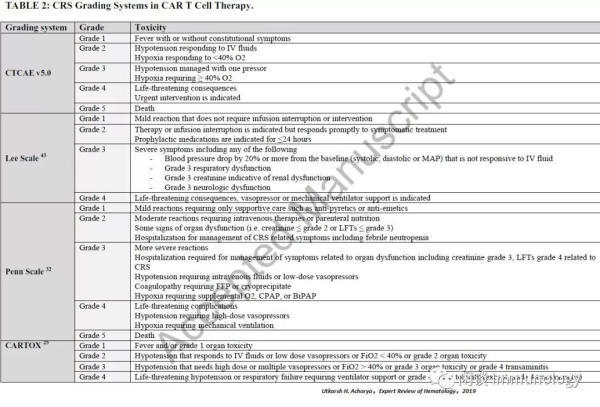
CRS分级
美国国家癌症研究所不良事件通用术语标准(CTCAE v4.0)没有充分描述CAR-T治疗相关CRS。因此,在CAR-T细胞治疗的初步试验中,使用了多个其他分级系统来描述毒性,并继续用于描述CAR-T相关CRS。CTCAE v5.0最近出版了,并且越来越多地被使用。尽管如此,以前使用的其他分级标准仍然有助于从不同角度描述CRS。目前使用的主要量表包括Lee标准、Penn量表和最近提出的CARTOX CRS标准。一般来说,定义毒性的一级:包括下表中所描述的非危及生命的体质症状。2级和3级:标准症状是可变的,依据不同程度的低血压和接受血管紧张素使用剂量。典型情况下,缺氧需要补充的FiO 2>40%和/或3级器官毒性和/或4级跨性鼻炎被认为是3级CRS。4级CRS包括危及生命的症状,需要机械通气支持或4级器官毒性(不包括经炎)。
CRS诊断相关生物标志物组合
其他因素如细胞因子或细胞因子受体基因多态性可能影响CRS的风险。临床试验表明,抗CD 19 CAR T细胞在体内诱导全身炎症反应,引起CRS。血清sIL-2Rα、IL-6、IFN-γ、IP 10、IL-5、IL-10、Flt-3L、GM-CSF等促炎细胞因子的升高在以往的研究中被认为是潜在的生物标志物。Teachi等人研究了51例包含39例儿童和12例成人复发或难治性B患者的细胞因子谱。4~5级CRS患者24种细胞因子,IL-6、IL-8、SIL-6R、MCP1和IFNγ的峰值水平明显高于0~3级CRS患者。早期观察发现重度CRS患者在CAR T细胞输注后第3天sgp 130和IFNγ水平明显升高,提示这些细胞因子可预测重度CRS后T细胞治疗的早期发病。值得注意的是,早期测量IL-6水平(前3天)未能区分4-5级和0-3级CRS。这很可能是因为IL-6水平没有升高,因此可能不作为预测标记。基于的在确定的细胞因子及其变化中,使用了16种预测模型。多种统计模型,如前瞻性logistic回归和决策树。组合队列的最佳模型使用了sIL1RA, IFNγ 和sgp130。SGP130的敏感性和特异性分别为86%和89%。不幸的是,SIL1RA和SGP130不容易获得测试,因此限制了使用。
在一项研究中,Hay等人对133名成人进行了回顾性、多变量分析。采用抗CD19 CART细胞治疗ALL、NHL和CLL的患者CRS。输注后36小时内4级CRS的患者显示浓度较高,包括IFN、IL-6、IL-8、IL-10、IL-15以及单核细胞趋化蛋白-1(MCP-1)、TNFRP55和MIP1R。此外,使用这些细胞因子的分类树建模在存在发热的情况下,血清MCP-1浓度为1343.5pg/ml可提高对4个CRS发展等级的患者的识别。总之,这些数据支持对细胞因子变化程度的理解,可用于预测抗CD19-CART细胞输注的患者的CRS严重程度。
与目前尚不普及的细胞因子检测方法相比,CRP的测定是一种快速、常规的方法,进一步强调了它在临床上的适用性。CRP是一种在IL-6作用下产生的急性期反应物,以往的研究表明CRP是许多炎症疾病中IL-6可靠的替代物。在Lee等人的一项研究中,CRP水平与IL-6水平呈正相关,严重CRS患者的IL-6峰值明显高于轻度或无CRS患者。同样,Davila等人。发现在所有患者中,CRP水平与CRS严重程度有关。此外,只有重度CRS患者才能观察到CRP≥20 mg/dl。相反,最近的一项研究表明,包括CRP、铁蛋白、LDH、AST、ALT、BUN和肌酐在内的标准临床实验室指标对预测严重CRS没有帮助,早期评估CRP不能预测严重或临床意义的CRS。然而,重要的是,CRP水平在感染患者中可能升高,并可能在疑似CRS中混淆感染的存在。因此,虽然CRP是一种常用的CRS评估工具,但不应单独使用,因为它可能无法识别出CRS的高危患者,而实验室价值的解释仍然需要纳入临床背景,以区分CRS病例和微生物感染病例。发现重度CRS具有非常高的铁蛋白,由于缺乏特异性,使用铁蛋白作为唯一的CRS生物标记物也受到限制。铁蛋白在全身炎症和铁过载的设置中也可升高。在患有晚期疾病的患者中经常观察到这一点。内皮的升高标记Angiopoietin-2和von Willebrand 因子的激活揭示了内皮激活可能与更严重的CRS相关联。
CRS临床管理
考虑到CAR-T细胞制造和产品中的异质性,利用细胞剂量、淋巴消耗,化疗和不同的患者人群CART细胞试验,建立标准化的预测方法和CRS的管理是困难的。CRS的管理通常涉及使用积极的支持性治疗,包括解热、镇痛、补充氧和静脉输液以及用于减弱免疫的直接措施,在中度至重度CRS病例中对治疗的反应。用于还原的试剂与T细胞扩增相关的免疫应答可包括针对的抗IL-6治疗(托珠单抗)和/或皮质类固醇。然而,托珠单抗是广泛的第一个支持性治疗后选择,因为皮质类固醇理论上可能影响抗肿瘤CAR-T细胞的作用。
tocilizumab是一种与IL-6受体(IL-6R)结合的人源化单克隆抗体(IgG 1κ),可用于治疗类风湿关节炎、幼年特发性关节炎和多关节青少年类风湿性关节炎。此外,tocilizumab对多种疾病均有疗效,包括克罗恩病、Castleman病和糖皮质激素难治性慢性移植物-VS-宿主病。tocilizumab可竞争性地抑制IL-6与受体结合的能力,阻断CAR-T细胞输注后诱导促炎反应的反式信号通路。IL6R阻断治疗严重全身CRS的症状和体征已被证明是非常有效的,而tocilizumab被FDA批准用于治疗CART细胞介导的CRS。在组织学临床试验中,37%的B-ALL和4%的NHL患者使用了tocilizumab。在紫杉醇的临床试验中,40%以上的大B细胞淋巴瘤患者接受了tocilizumab。tocilizumab一般耐受性好;然而,给药后IL-6水平的生化升高已经被观察到,并且在使用几天后,神经毒性被发现延迟。根据类风湿关节炎患者的经验,tocilizumab可能增加机会性感染的风险,如病毒、真菌和分枝杆菌感染,特别是慢性使用。
皮质类固醇也是治疗严重系统性CRS的主要部分。然而,它们的反应通常比tocilizumab延迟得多,理论上担心,如果不加选择地使用,它们的淋巴毒性可能会损害CAR T细胞的效能。然而,在Zuma-1使用抗CD 19定向CAR T细胞治疗难治性DLBCL中,没有发现使用它的负面影响。尽管如此,皮质类固醇被认为是一种补充IL-6阻断的辅助措施,对于高级别的系统性CRS患者,对tocilizumab没有反应。治疗紫杉醇后的CRS管理建议包括每日两次使用甲基强的松龙1mg/kg,或每6小时使用地塞米松10毫克,3级CRS使用tocilizumab。对于4级CRS,甲基强的松1000 mg,连续3天。另一方面,在使用甲基强的松2mg/kg接受情况下,如在tocilizumab治疗后12至18小时内没有任何改善,则推荐使用糖皮质激素。
其他潜在目标免疫抑制剂包括英夫利昔单抗、依那西普、Anakinra和环磷胺,Sylvant(siltuximab) 等,也在理论上有一定效果。不幸的是,所有这些试剂的给药和使用都是根据其在风湿病学文献中的批准推断。有这些经验的经验与托珠单抗或皮质类固醇相比,是有限的。

来源:闲谈 Immunology 作者:挑食的免疫喵

来源:网络
版权及免责声明:本网站所有文章除标明原创外,均来自网络。登载本文的目的为传播行业信息,内容仅供参考,如有侵权请联系答魔删除。文章版权归原作者及原出处所有。本网拥有对此声明的最终解释权。
{replyUser1} 回复 {replyUser2}:{content}