CRISPR-Cas9在治疗β-地中海贫血疾病中的应用
一、β-地中海贫血的形成原理
CRISPR-Cas9不仅是基因功能调控的主力工具,也是疾病治疗中的良好帮手。在此我们先了解一下β-地中海贫血(β-thalassemia)的形成原理。众所周知,血红蛋白是红细胞内一种负责运输氧气的特殊蛋白质,由血红素和珠蛋白构成。珠蛋白是一种四聚体,由两对不同的珠蛋白链(α链和β链)组成。
β-地中海贫血是人β-珠蛋白(β- globin)基因点突变或缺失引起的常染色体隐性遗传病,因β-珠蛋白完全不能合成或仅能部分合成引起α、β链合成比例失衡,导致不稳定的游离型α-珠蛋白堆积、变性及降解,从而产生细胞毒性活性氧化物,损害红细胞的成熟和活力,破坏红细胞膜,最终导致溶血性贫血。
α链和β链的数量不平衡是导致地中海贫血的直接原因,β-地中海贫血患者的β链合成减少或缺失会导致α链相对过剩,减少α链合成可缓解地中海贫血的疾病症状。对α-珠蛋白的顺式调控元件进行基因编辑,可调控α-珠蛋白的表达量。研究表明利用CRISPR-Cas9敲除β-地中海贫血患者造血干细胞中α-珠蛋白基因增强子MCS-R2的核心元件,可降低α链的合成水平。
图1正常红细胞与β-地中海贫血红细胞对比图
据报道,全球有8000万~9000万人(约占全球总人口的1.5 %)为β-地中海贫血携带者。β-地中海贫血好发于地中海地区,故因此得名“地中海贫血”。其次地中海贫血为中东、印度、巴基斯坦,是我国南方常见的遗传性血液病之一,患病率达2.21 %,其中广西、广东、海南、贵州等地区也是是地贫高发区,携带率分别约为4.8 %、2.54 %、2.27 %、3.23 %。
β-地中海贫血发病的分子机制复杂多样,其致病基因HBB位于第11号染色体,到目前为止已经确认了200多个致病突变类型,其中大多数是单核苷酸替换、缺失或寡核苷酸插入导致移码。在中国4种常见突变类型为CD41/42(-CTTT)碱基缺失,CD17(AAG>TAG)、IVS2-654(C>T)和TATAbox28(A>G)点突变,约占全部病例的90 %,尤以CD41/42(-CTTT)基因型最多。根据临床症状的严重程度,β-地中海贫血可分为轻型、中间型、重型三类,HBB基因的突变类型决定了β-珠蛋白合成抑制的程度,从而决定了临床症状的严重程度。当患者为重型β-地中海贫血患者时,出生后一年内即可观察出病征,其特殊表现有:头大、眼距增宽、马鞍鼻、前额突出、两颊突出,其典型表现为臀状头,长骨可骨折。骨骼改变是骨髓造血功能亢进、骨髓腔变宽、皮质变薄所致。少数患者在肋骨及脊椎之间发生胸腔肿块,亦可见胆石症、下肢溃疡。常见并发症有急性心包炎、继发性脾功能亢进、继发性血色病。
星耀小TIP:β-地中海贫血的三类症状体现:轻型:轻度贫血或无症状,一般在调查家族史时发现。中间型:轻度至中度贫血,患者大多可存活至成年。重型:出生数日即出现贫血、肝脾肿大进行性加重,黄疸,并有发育不良,患者可伴有肺动脉高压、血栓形成、腿部溃疡、骨质疏松症、自身免疫性溶血性贫血等严重并发症。
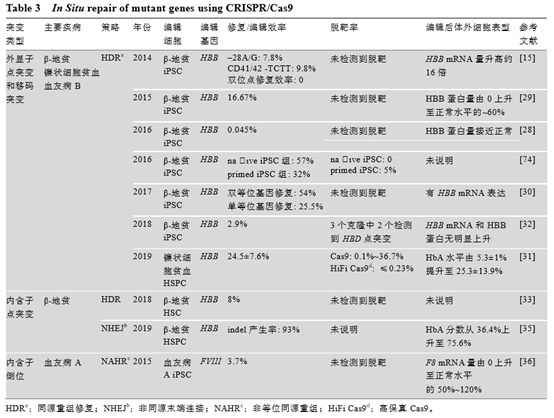
外显子点突变/移码突变可能是导致蛋白质的结构或合成数量异常,如β-地中海贫血,此时便需要精确修复才能恢复基因功能。常用的方法为基因同源重组修复途径,但造血干细胞(hematopoietic stem cells,HSCs)和诱导多能干细胞(induced pluripotent stem cells,iPSCs)中同源重组的效率低且不稳定,所以产生了另一种精确修复的方法,即使用CRISPR-Cas9对基因组的突变进行修复,相交同源重组技术效率更高,稳定性更高。
图2 CRISPR-Cas9原位修复突变基因的研究
二、CRISPR-Cas9在β-地中海贫血中的应用
目前异体造血干细胞移植是根治β-地贫的唯一手段,但与患者组织相容性抗原匹配的供体难寻。骨髓造血干细胞是血液系统中的成体干细胞,可分化产生各系造血祖细胞(hemato-poietic progenitor cells, HPCs),进而分化为各系血细胞,为造血系统提供新的细胞来源,同时造血干细胞不断自我增殖以维持自身数量的相对稳定。
研究表明,CD34是造血干/祖细胞(HSPCs)的特异性分子标志,并在HSPCs归巢过程中发挥重要作用。诱导多能性干细胞是通过体细胞重编程技术从成体细胞获得的类似于胚胎干细胞(embryonic stem cells,ESCs)的多能性干细胞。iPSCs来源于病人本身的体细胞,易于获取,不会发生免疫排斥反应,并且具有类似ESCs的自我更新和多向分化潜能。因此,在CRISPR-Cas9基因编辑技术治疗β-地贫的相关研究中常使用CD341 HSPCs和iPSCs作为疾病模型建立、基因治疗和细胞治疗的重要靶细胞。
三、建立β-地中海贫血体内外模型
β-地中海贫血体内外使用的是小鼠模型。小鼠有与人类相近的基因序列,以及成本低、繁殖快的优点,目前许多实验都将小鼠作为首选的实验动物,且动物模型的建立对生物医学研究和探寻疾病发生的发展进程中有着不可或缺的作用。
小鼠β-珠蛋白基因的缺失会导致与人类β-地中海贫血相似的表型,可推断出β-珠蛋白在小鼠和人类之间结构与功能的保守性,故早在20多年前就已有利用传统的同源重组技术敲除β-地贫相关基因的敲除鼠,具体的同源重组技术流程可参考耀海生物的公众号“耀海人才”的往期文章《大肠杆菌Red同源重组原理》、《大肠杆菌Red同源重组敲除—感受态细胞的制备》、《大肠杆菌Red同源重组-引物设计及其应用》。
图3 CRIPSPR-Cas9 基因编辑原理
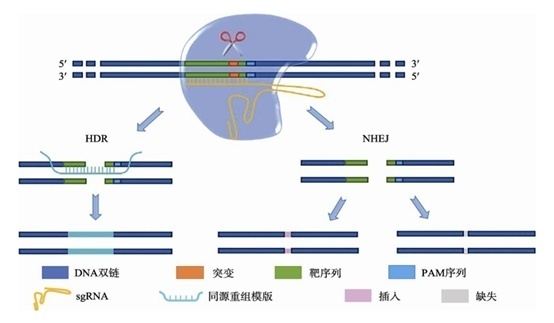
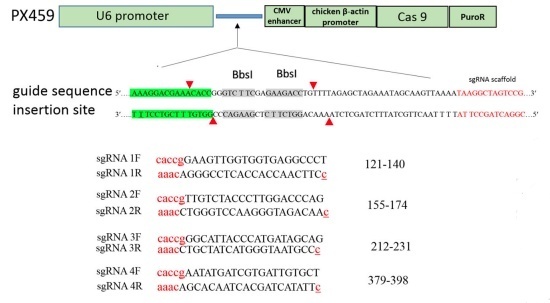
新兴的CRISPR-Cas9技术使得构建各种β-地中海贫血模型更快捷方便。具体方法如下:使用px459载体,该载体可以表达SpCas9、一个针对Hbb‐b1设计而成的gRNA和一个嘌呤霉素抗性基因。运用CRISPR-Cas9技术对载体基因进行编辑,使其可以定向敲除β-珠蛋白合成的基因。
图4 载体构建
选用小鼠胚胎干细胞(ES‐E14TG2a)进行转染,转染48小时后,通过添加最佳的嘌呤霉素浓度进行转染细胞选择,经过筛选得到HBB基因敲除的小鼠单克隆ES细胞。
对小鼠胚胎干细胞进行进一步的筛选,测定基因序列以选择我们所需要的小鼠胚胎干细胞并进行体外红细胞分化,第一步为初级分化,将ES细胞以单细胞形式悬浮细胞培养基中,以悬滴形式悬浮48小时;第二步,将第一步培养的细胞至于特定的甲基纤维素基培养基培养10~15天,确定造血集落的数量和类型。制备成β-地中海贫血小鼠的红细胞模型。
通过直观模型的建立,对于我们研究、治疗β-地中海贫血具有深远意义,模型方法是生物工程中广泛使用的研究方法,在科学研究中具有重要作用。小鼠模型是基因治疗和基因编辑常用的动物模型,且近年来小鼠模型所展现出的众多优势使其在动物模型中独占鳌头,已经成为建立人类相关疾病模型和药物临床前评价研究等最为重要的动物模式。
四、β-地中海贫血治疗的临床应用
图5 CRISPR/Cas9 用于遗传性血液病基因治疗的研究
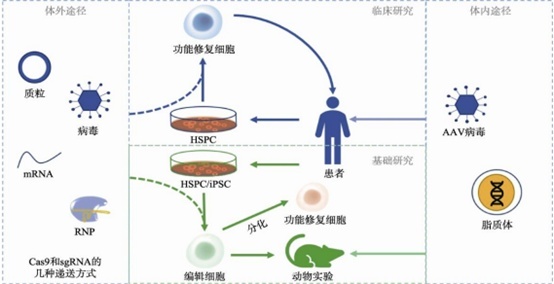
作为新一代的基因编辑工具,CRISPR-Cas9在治疗基因缺陷导致的疾病诸如β-地中海贫血中展现出了极大的优势。主要可以通过校正病人β-珠蛋白基因(HBB)基因原位突变、调控γ-珠蛋白基因表达、抑制α-珠蛋白基因(HBA)基因表达来从根本上对患者进行治愈。
日前,中南大学湘雅医院付斌等人通过CRISPR基因编辑技术重激活γ珠蛋白治疗β0/β0重度地中海贫血儿童的研究获得成功,该研究先从患者体内提取造血干细胞,在实验室重新激活胎儿期正常、出生后沉默的γ基因,通过表达患者自身的胎儿血红蛋白,让它代替β基因来行使携氧功能,再将激活后的干细胞输回患者体内,让其生成正常的血细胞。本研究激活γ基因的重激活效率已超过90%,保证了稳定持久的临床疗效。在基因编辑过程未使用病毒作为载体,有效避免了病毒带来的潜在的基因治疗风险。
星耀小TIP:重型地中海贫血的常规治疗方法是规范性终身输血和去铁治疗,不仅疗效有限并往往发生铁过载,患者的工作、生活和学习存在严重障碍,而且存在输血困难、严重消耗血资源、社会医疗成本高昂等限制性因素。因此在基因层面解决β-地中海贫血已成为当下研究的热点。
五、总结
通过上述介绍,我们可以知道CRISPR-Cas9相交传统的同源重组技术,在治疗β-地中海贫血疾病中具有更为有效且便捷的作用。CRISPR-Cas9技术可进行信号通路相关基因的寻找,药物靶点筛选,药物原发性研究及基因治疗等,可以预见CRISPR0-Cas9在未来的道路上将继续飞速发展,广泛应用于生物工程中。
来源:

来源:网络
版权及免责声明:本网站所有文章除标明原创外,均来自网络。登载本文的目的为传播行业信息,内容仅供参考,如有侵权请联系答魔删除。文章版权归原作者及原出处所有。本网拥有对此声明的最终解释权。
{replyUser1} 回复 {replyUser2}:{content}