新型偶联药物浅析
化疗是癌症的重要治疗手段之一。但是化疗药物对肿瘤细胞和正常细胞无特异识别功能,会导致严重的全身系统性毒性。通过癌细胞表面表达的分子选择性结合并发挥作用的靶向治疗是癌症治疗的一个重大进展,因为与传统的细胞毒性药物相比,靶向治疗具有更高的疗效和更好的耐受性。
近年来,ADC(antibody-drug conjugate)药物研发如火如荼,国内外许多药企布局ADC赛道,当下ADC药物已成为国内外肿瘤靶向治疗领域不可忽视的抗癌药物。ADC的成功,推动了偶联药物开发的热情。一些新型的偶联药物类型不断涌现,如:PDC(多肽偶联药物)、RDC(核素偶联药物)、SMDC(小分子偶联药物)、AOC(抗体寡核苷酸偶联物)、ADeC(抗体降解偶联药物)等。与ADC药物相比,PDC药物则具有分子量小、肿瘤穿透性强、免疫原性低以及生产成本低等优点,有望成为继小分子药物、单克隆抗体、ADC药物之后新一代靶向抗癌药物。
倚锋资本将通过浅析偶联药物技术手段、盘点各类偶联药物、梳理行业代表性企业,带大家一览新型偶联药物行业的前沿进展和潜在机会。
01 PDC药物介绍
偶联药物结构
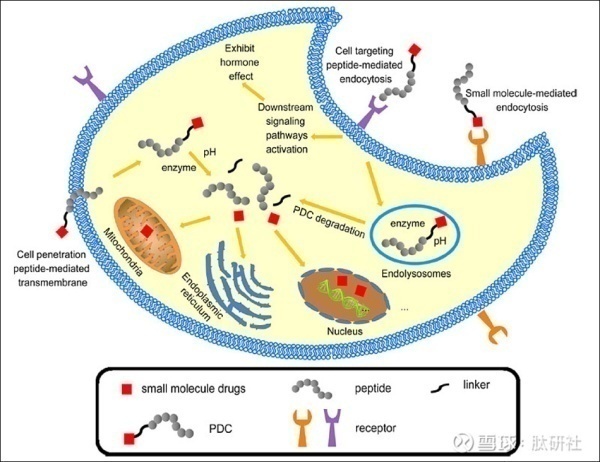
为靶向给药而开发的偶联药物通常由三种成分组成,所有这些成分都有助于药物的整体生物疗效和选择性。载体部分包括在该结构中,其靶向肿瘤特异性标记物。除多肽外,还研究了一系列其他小分子和生物制剂,如天然蛋白质、抗体、粘附体、设计的锚蛋白重复蛋白和适配体,以提供肿瘤选择性。
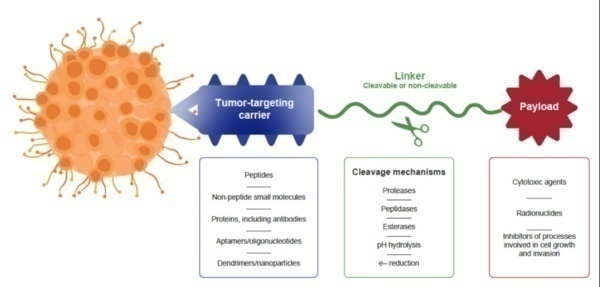
PDC定义
多肽偶联药物(PDC)由靶向肽、细胞毒性药物和连接子(Linker)三部分构成,是将靶向肽作为靶 向给药载体,与毒性药物分子共价偶联,来增强药物的靶向性,其中连接子的选择对于PDC药物能 否稳定达到靶向部位具有重要的影响。PDC的目标是为了提高化疗药物的疗效,克服化疗药物的循环半衰期短和脱靶副作用的挑战。
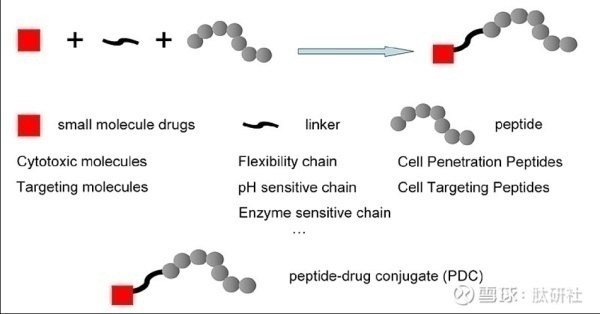
PDC vs ADC
凭借分子量小、肿瘤穿透性强等特点,PDC更容易在实体瘤中发挥作用。相比较而言,ADC由于抗体 的分子量较大,限制了穿透实体肿瘤的能力。近年来ADC获批的适应症大多是针对血液恶性肿瘤,实体瘤中较多的只有乳腺癌。
不仅如此,PDC的细胞毒性药物选择面更广泛。由于较强的肿瘤组织渗透性,PDC药物能够在靶标处 累积达到高浓度,从而高效地杀伤肿瘤细胞。因此PDC可以选择阿霉素、紫杉醇等毒性相对较低且普 遍应用于临床的化疗药物,作为PDC的毒性弹头,载药量也更高。
而ADC虽可以利用抗体的精准靶向,将“有毒弹头”选择性递送至癌细胞。但受抗体大分子的限制, 在肿瘤组织的渗透率极低,只能选择高毒性的化疗药物,例如赫赛莱的化疗药物DM1、维布妥昔单抗的细胞毒剂单甲基阿司他丁E(MMAE),才能保证在低剂量下的肿瘤杀伤效果。
此外,PDC药物的设计与生产上更为简单,成本也较低。

ADC市场一直是快节奏的,而PDC的情况并非如此。一个重要的因素是,多肽的由于其相对较小的尺寸,它 们对肾脏的清除速度很快,口服生物利用度极其有限。这个问题已经通过各种方法来解决,例如化学修饰和物理技术(环化、化学修饰、制剂优化) 及通过纳米材料增强PDC的PK特性。这些已建立的方法已被证明可以 通过改善细胞通透性、增强化学和蛋白水解稳定性、降低肾脏整体清除率等途径来改善多肽的ADME性质,从而延长循环半衰期。这为PDC进入临床试验提供了极大的潜力。
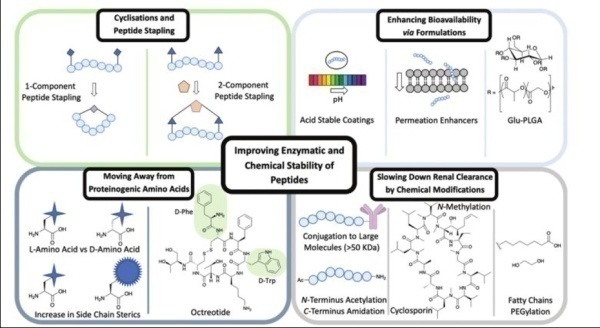
PDC药物优势
1) 传统的肿瘤药物是疏水性的,而用赋形剂溶解不溶性药物可能引入新的问题:具有自身毒性的添加溶剂。癌症药物紫杉醇溶解性非常不好,Taxol就用表面活性剂Cremophor EL溶解药物来注射,这导致严重的过敏反应和神经毒性。而将肿瘤药物结合在亲水的肽链上能够减少药物溶解的问题。
2) 传统的化疗药物对肿瘤杀伤的同时也会损伤正常组织。有效的方法就是考虑正常组织和肿瘤细胞表面受体的差别。靶向细胞表面受体的肽链设计就能够特异传输药物的特定的肿瘤细胞上。Integrin肽链被广泛应用,因为它们对于组织的生理发育,维持和修复是必需的,并且在各种疾病,特别是癌症的病理过程中具有突出特征;而且Integrin携带的药物似乎对肝脏和心脏的毒副作用更小。
3) 传统的药物进入细胞膜内化能力是个棘手的问题。早期的研究聚焦在研发一个合适的亲水-疏水平衡从而使药物穿过去极化的细胞膜,这是一个非常耗时且吃力不讨好的工作。比如说,环磷酰胺CsA对皮肤疾病效果非常好,但是系统性输送药物会出现脱靶的毒副作用。本身,CsA没有皮肤穿透性,为了克服了这个问题,有人通过pH敏感连接子将精氨酸的七聚体与CsA缀合,产生了R7-CsA。R7-CsA能够有效的在皮肤细胞中进行转运,从而到达皮肤T淋巴细胞抑制皮肤炎症。环磷酰胺的例子代表了现行通用的增强不易吸收药物体内运输的策略。
4) 用于化疗的单一药物由于靶向癌细胞中的药物阻力而常常无法根除所有癌细胞。因此,非交叉抗癌药物被广泛用于有效的癌症治疗。
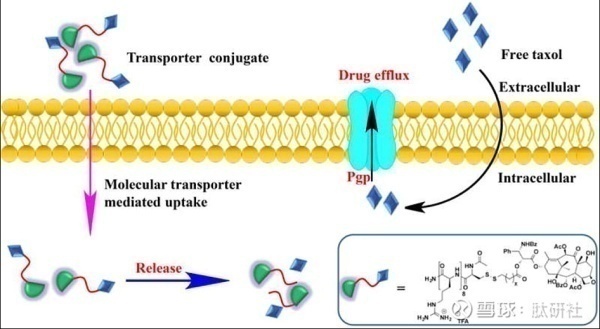
5) 传统的肿瘤药物会出现耐药性。耐药性是许多肿瘤化疗药物失败的主要原因,特别是在复发病人治疗上。肿瘤的耐药机制是增加了介导单向能量依赖药物传输的膜蛋白的表达。这种蛋白可以拦截并且吐出药物。这种蛋白对一种药物的抵抗,常常也会对其他药物的抵抗。富含Guanidinium的PDCs可以帮助规避这些外排蛋白 ,因为它们的内化不涉及穿过膜的被动扩散(药物进入细胞的机制),因此可以避免膜蛋白介导的释放。
PDC药物设计原则
1) PDC中包含的肽必须选择性结合并以最佳亲和力与特定的受体结合,该受体存在于目标组织的细胞表面,而不是其胞质或细胞核内(即类固醇受体)。
2) 所选受体应在癌细胞上独特表达或过表达(通常是正常细胞的3倍或更高)。
3) 肽载体应以与药物或/和荧光团缀合可行的方式构建。在固相肽合成过程中,通常通过正交偶联在赖氨酸,半胱氨酸和谷氨酸上或在肽的游离N端发生缀合。但是,应仔细选择缀合位点,因为在肽结构微环境中诱导的扰动可能会导致其与靶向受体的结合亲和力/选择性消失。
4) 应该仔细选择Linker,以使PDC达到最佳性能。错误的选择可能导致肽与受体的结合亲和力降低或/和药物治疗窗口的减少。另外,它应在血液循环中在酶学上稳定,以便有效地到达恶性肿瘤部位并在其微环境中释放有效载荷,从而降低脱靶毒性。
5) 细胞毒剂应包含可以与肽链相连的适当官能团,如果不存在,则应考虑合理安装细胞毒剂的最终衍生物,以保持最初的细胞毒性的活性。
02 PDC药物组分
细胞毒剂
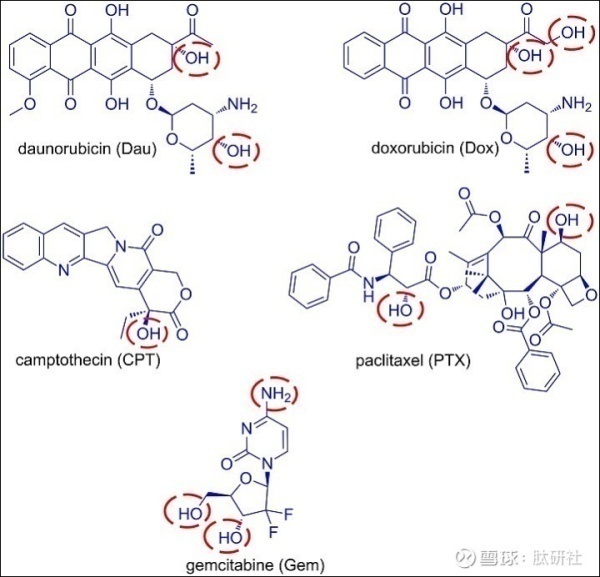
根据国家癌症研究所的数据,目前有250多种FDA批准的抗癌药物用于治疗恶性肿瘤。在这种大量的细胞毒性药物中 ,其中一些已被用作PDC中的有毒弹头,五个代表性的例子是吉西他滨,阿霉素,柔红霉素,紫杉醇和喜树碱。
这些原始抗癌药的主要缺点是其不受控制的毒性,导致严重的副作用。如果不添加靶向部分,它们将癌细胞与正常细胞区分开的能力很低。此外,添加肽作为靶向载体可以增强母体细胞毒剂的药代动力学和治疗窗口。由于不同的药物可能采用不同的机制杀死细胞,因此根据表征目标癌细胞的特征选择合适的药物。
Linker选择
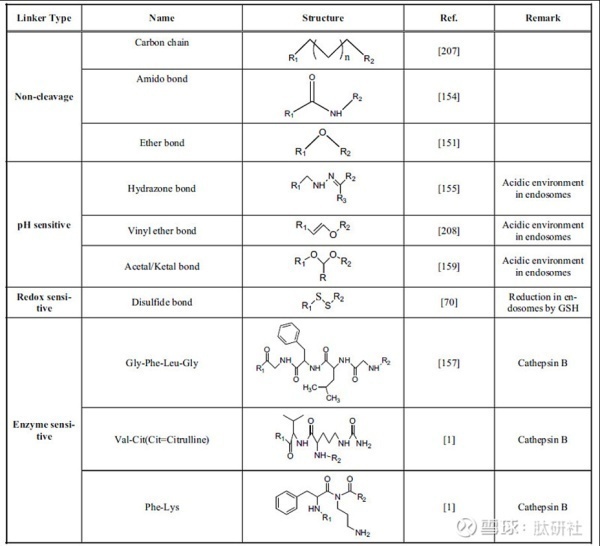
在PDC设计过程中应考虑的另一个关键方面是将肽和药物束缚在一起的Linker。连接体必须精心设计,以免干扰肽与其受体的结合亲和力和药物功效。不合适的Linker可能会阻碍药物从PDC释放,因此会降低其总体治疗效力。PDC中使用的Linker存在不同的类别,并且它们的长度,稳定性,释放机理,官能团,亲水性/疏水性等不同。
选择连接体(Linker)是为了允许足够的循环时间,使药物到达其目标细胞。因此,PDC应足够稳定,使肽 、连接物和药物在到达靶细胞之前不会被裂解或代谢,并使足够浓度的PDC到达靶细胞,从而提高药物杀灭肿瘤的效果。大多数连接体在系统中就开始被裂解,从血浆(血液)开始,然后是癌细胞的细胞外环境。连接区域常见的官能团大致可分为四类:酶可裂解(酯、酰胺和氨基甲酸酯)、酸可裂解(肼和碳酸盐酯)、可还原二硫醚和不可裂解(硫醚、肟和三唑)。
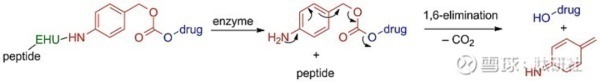
近年来,PDC的Linker中另一个迅速兴起的类别是自焚或自毁性间隔子/Linker。这种类型的Linker/间隔物提供了在同时发生级联反应后释放活性药物的能力,对氨基苯甲醇(PABC;红色)是一个代表性的例子,可以 通过酰胺键在氨基上与酶水解单元(EHU;绿色)和肿瘤靶向元件(即肿瘤归巢肽;黑色)相对位置的醇基可以通过碳酸酯/氨基甲酸酯键与细胞毒剂(蓝色)相连。EHU被设计为在目标肿瘤微环境(即组织蛋白酶B) 中过表达的蛋白酶的底物。一旦EHU被这些酶识别,它就会被裂解,从而导致活性药物通过快速级联反应释放出来。
常见Linker
肽链
PDC中使用的肽分为两类:细胞穿透肽(CPPs )和细胞靶向肽(CTPs)。具有穿透细胞归巢肽的PDC通过非特异性机制进入细胞,而具有细胞靶向肽的PDC通过特异 性结合肿瘤细胞表面的抗原或受体,以介导细胞毒性有效载荷进入肿瘤细胞。CPPs由于其低细胞特异性,这些类型的PDC的应用受到限制。相反,CTP表现出与单克隆抗体类似的作用,同时克服了单克隆抗体的某些缺点,因而得到广泛应用。
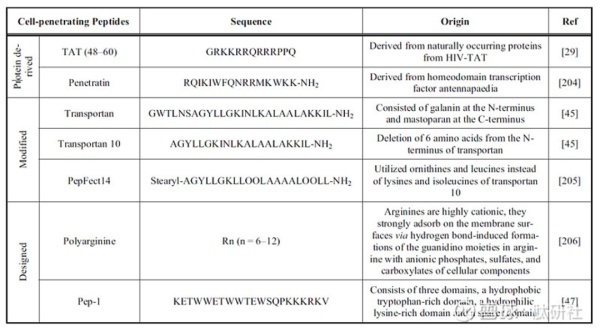
CPPs
细胞穿透肽是短肽(少于30个残基),能够穿过细胞膜。CPPs的共同特征是正电荷和两亲性。CPPs可分为三种类别 :蛋白质源性CPPs,修饰CPPs和设计CPPs。
CTPs
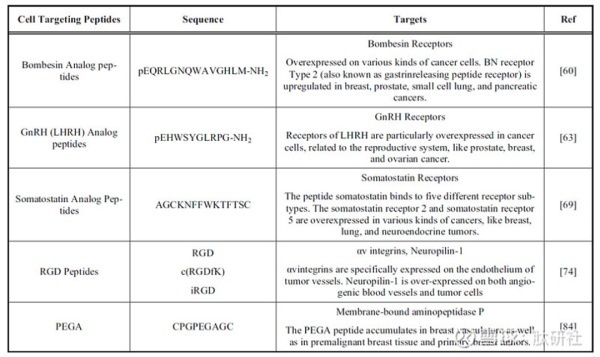
目前,具有特殊结合能力,能够与特定细胞和组织进行特异化结合的多肽已经有广泛的报道。这些肽(称为细胞靶向 肽,CTPs)能够与特定细胞内过度表达的受体相互作用。
03 PDC已上市药物
PDC药物适应症
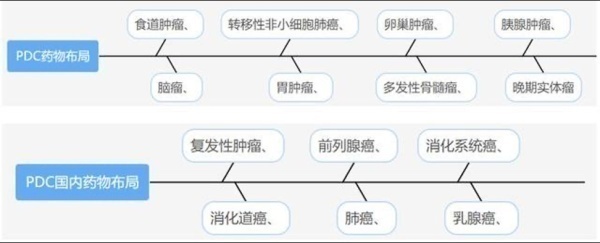
全球代表性PDC药物研发的适应症包含种类丰富,主要有食道肿瘤、脑瘤、转移性非小细胞肺癌、胃肿瘤 、卵巢肿瘤、多发性骨髓瘤、胰腺肿瘤、晚期实体瘤等。国内在研PDC药物适应症主要有复发性肿瘤、消化道癌、前列腺癌、肺癌、消化系统癌、乳腺癌等。由此看出PDC药物适应症种类繁多,未来市场规模庞大,行业发展前景较好。
已上市PDC药物
目前PDC领域尚处于开发的洼地,国内研究公司并不多见。全球也仅有2款PDC药物上市,分别是2018年获批的Lutathera,以及近期获得FDA加速批准的melflufen,由Oncopeptipes公司开发。Lutathera由诺华子公司Advanced Accelerator Applications S.A开发上市,是第一款肽受体放射性核素治疗( PRRT)药物。Melflufen的加速获批是得益于一项关键II期HORIZON的临床结果。试验纳入157例患者与地塞米松联用,治疗复发/难治性多发性骨髓瘤(MM),总缓解率达到23.7%,中位缓解持续时间为4.2个月。

Melflufen
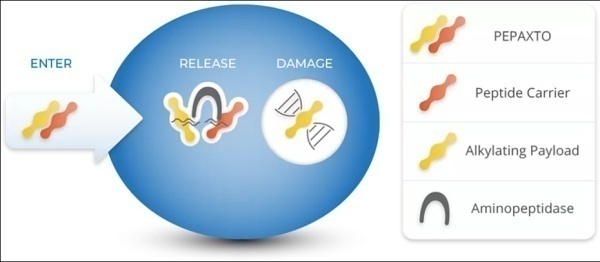
Melflufen是一款靶向氨肽酶(aminopeptidase)的First-in-Class PDC药物。由于高亲脂性,Pepaxto能被骨髓瘤细胞迅速吸收。进入细胞内,Pepaxto的偶联肽会立即被氨肽酶裂解,释放出亲水性烷化剂的有效荷载马法兰,以对抗过度表达且可以释放毒性的氨肽酶。基于一项II期关键临床研究HORIZON,2020年2月26日,FDA 基于一项II期关键临床研究HORIZON,FDA加速批准Pepaxto上市(OCEAN为上市后的验证性临床研究),与地塞米松联用于治疗三重难治性/复发性多发性骨髓瘤(R/R MM)。
但在获批上市没多久(2021年7月28日),Oncopeptides就报告了来自Pepaxto验证性临床试验OCEAN死亡率增高的不好消息。临床发现,与接受泊马度胺和地塞米松治疗的患者相比,接受Pepaxto+地塞米松治疗的患者死亡人数更多,Pepaxto的死亡风险高出10%。为此,FDA很快向Pepaxto发出警告,要求公司暂停该临床试验以及其他正在进行的临床试验入组。
不过,2021年9月第18届国际骨髓瘤研讨会(IMW)上公布的III期OCEAN研究最新数据似乎给Oncopeptides带来了重返市场的信心。试验结果显示(据独立审查委员会评估),OCEAN达到了无进展生存期(PFS)的主要终点;Pepaxto组的PFS中位数为6.8个月,泊马度胺组为4.9个月,危险比(HR)为0.79。在意向治疗(ITT) 人群是关键次要终点,总生存期(OS)有利于泊马度胺,HR为1.10;Pepaxto组总缓解率(ORR)为33%,泊马度胺组为27%。
目前,基于这个临床数据,Oncopeptides重新考虑此前自愿撤市的决定,并与FDA密切沟通,以重新审查临床数据。
04 PDC在研药物
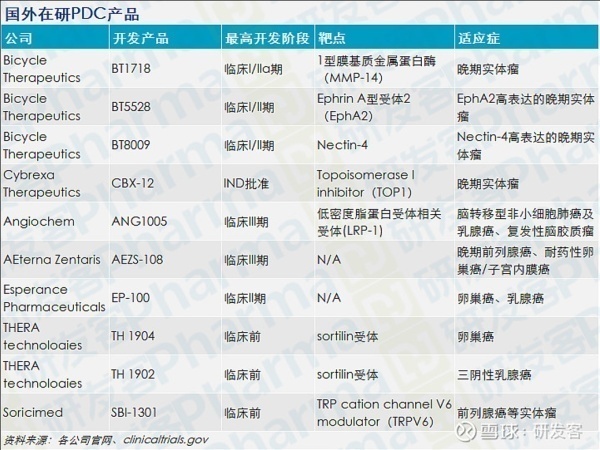
国外临床产品
国外已有7款产品进入临床。
PDC的在研产品以国外公司为主,研发进展较快且活跃的公司有Bicycle Therapeutics、Cybrexa Therapeutics等。
国外开发技术
提高多肽稳定性的方法之一是环化技术。如Bicycle公司名称一样,该公司将PDC药物中的多肽设计成 Bicycle,以稳定的双环肽分子结构与靶蛋白结合,达到与抗体类似的高亲和力和靶向选择性。
而Cybrexa公司的CBX-12,是依托alphalexTM技术平台开发而来。该技术由pHLIP®肽、连接子和小分子 抗癌剂组成,其中pHLIP®肽最早由耶鲁大学和罗德岛大学开发,并独家授权给pHLIP,后Cybrexa与 pHLIP形成合作伙伴关系,拥有pHLIP®肽的全球独家开发和商业化权利。
Bicycle Therapeutics
该公司独创地将多肽偶联药物中的多肽设计成Bicycle,它是由短的线形肽在“支架”的作用下形成两个环的 短肽。Bicycle的长度一般在9-20个氨基酸之间,序列中有3个半胱氨酸残基。这些半胱氨酸残基与小分子连 接物反应,将肽限制在刚性构象中。Bicycle的分子量在1.5~2KDa,具有良好的组织渗透率和肾脏清除率、 高度亲和力和选择性的优点,较大的分子足迹可使得蛋白间的相互作用具有靶向性,结合了生物药的药理学 和小分子药物的药动学的优势,没有免疫原性。
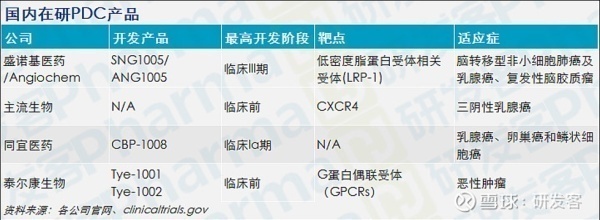
国内临床产品
由国内公司盛诺基医药与加拿大公司Angiochem合作开发的SNG1005,正在中国开展III期临床试验。是国内进展最快的PDC项目。
SNG1005是一款穿透血脑的多肽药物偶联物,将紫杉醇与氨基酸短肽偶联得到,能够将紫杉醇特异性递送至脑部 。目前Angiochem已完成了II期临床,结果显示,SNG1005在治疗乳腺癌脑软膜转移癌和复发性乳腺癌脑实质转移癌中,都显示出积极的临床治疗效果。其中在对乳腺癌脑转移患者颅内和颅外的治疗中,SNG1005的临床获益率分别达到了77%和86%。
国内代表企业
其他布局PDC产品的公司都是初创公司。其中同宜医药凭借核心技术平台BESTTM,开发出一系列抗肿瘤偶联药物(XDC)。其中以双靶向-配体偶联体CBP-1008进展最快,目前正在进行Ia期临床试验。
泰尔康生物和主流生物的PDC产品还都在临床前。泰尔康生物主要开发以GPCRs为靶点的PDC类药物,目前正在寻求天使轮融资,用以支持Tye-1001和Tye-1002两款产品的临床开发。
主流生物医药公司也率先布局了PDC赛道,开发靶向CXCR4的多肽偶联药物。公司总裁李风翔在2020 Think Big生物医药创新项目路演活动上介绍,主打产品MB1707有望在今年启动中美临床试验,另一款产品MB010 主要拓展和PD-1/ PD-L1联合用药的治疗方案。
临床代表性PDC药物
1.CBX-12
在CBX-12是由Cybrexa Therapeutics公司开发,该公司成立于2017年,是一家私营生物科技公司,致力于利用其 alphalexTM平台开发新一代肿瘤靶向癌症疗法。公司的先导化合物CBX-12是一种alphalexTM偶联物,包括强效拓扑异构酶I 抑制剂依喜替康。CBX-12是根据其突破性功效和临床前研究中证明的安全性来选择的。CBX-12与PD-1抑制剂和其他免疫肿瘤药物具有潜在的协同作用,该化合物有望在2021年进入晚期实体瘤的I/II期临床试验。此外,Cybrexa还在进行其他临床前毒素结合物研究项目以及合成致死研究项目,包括CBX-13,CBX-14和CBX-11。Cybrexa和大型制药公司的合作项目也在推进中。
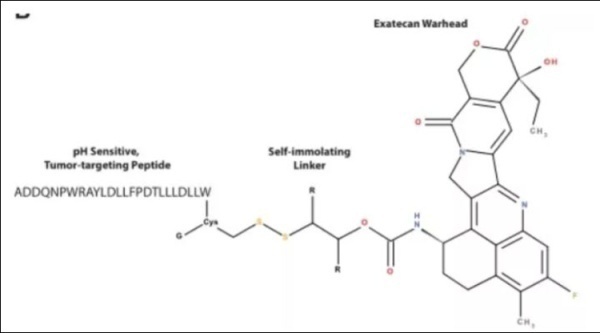
alphalexTM为肿瘤靶向技术平台,能够使抗癌剂在低pH条件下穿透癌细胞膜,并在酸性肿瘤微环境中于肿瘤细胞内选择性释放药物分子,同时最大限度降低对健康组织的毒性。该技术由pHLIP®肽、连接子和小分子抗癌剂(效 应分子)组成,实现了不依赖肿瘤抗原的靶向性和高效抗癌剂的细胞内传递,创造可以彻底改变治疗标准的疗法。
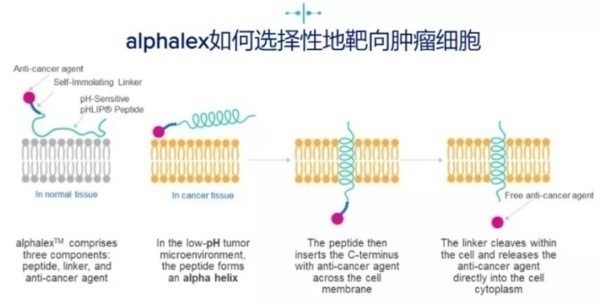
pHLIP®肽是一类针对酸性细胞表面的pH值敏感型插入肽,在低pH环境下会形成α螺旋,接着其连接抗癌剂的C- 末端会穿过细胞膜并释放毒性分子。该技术由耶鲁大学和罗德岛大学开发,并独家授权给pHLIP, Inc,该公司的任务则是将pHLIP®技术商业化。Cybrexa作为pHLIP的合作伙伴,拥有pHLIP®肽的全球独家开发和商业化权利,可用于研发包括DNA损伤修复抑制剂和细胞毒性分子在内的肿瘤治疗剂。pHLIP家族肽在其主要序列中具有相同的特征,并表现出相同的作用机制。这些共同特征主要包括:N端区域(序列 1)包含3-20个残基,主要由增加肽整体溶解度的极性氨基酸组成;中间区域(跨膜序列)包括15-25个残基不等,主要由疏水残基组成,但也包括在生理pH下带负电但在低pH下由于质子化而带中性电荷的氨基酸;以及一个包含0-10个残基的C端区域。
在血浆体外稳定性测定结果中,CBX-12显示良好的稳定性,在8小时内未检测到毒素的释放,在24小时时毒素释放极少,验证了CBX-12在血液循环中稳定性。在异种移植的动物模型中,也验证了CBX-12的血液循环稳定性和肿瘤细胞选择性。
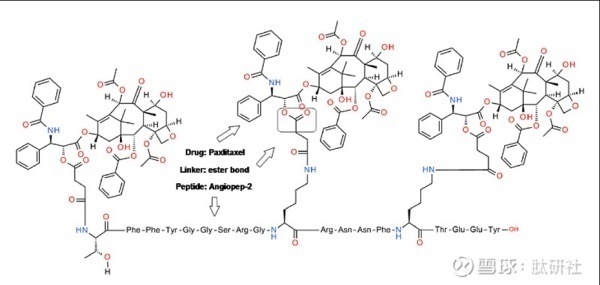
2. Angiopep-2-Paxlitaxel缀合物(ANG1005、GRN1005)
ANG1005(也称为GRN1005)是由angiopep-2和紫杉醇组成的药物结合物,旨在通过LPR1介导的胞吞作用增加大脑转运。该ANG1005缀合物包括相对紫杉醇的多达三个摩尔当量的肽意图最大限度增加细胞毒性药物浓度。
ANG1005对胶质母细胞瘤,肺癌和卵巢癌细胞具有优异的细胞毒性。此外,ANG1005以LPR1依赖的方式有效转运至大脑,并且不受P-gp外流的影响,否则会损害治疗效果。ANG1005显着延长了带有异种移植胶质母细胞瘤或肺癌细胞的小鼠的存活期。在复发性恶性神经胶质瘤肿瘤的1期临床试验中,ANG1005表现出3.6h的血浆半衰期,并且具有与紫杉醇相似的毒性,因此被很好地耐受。重要的是,ANG1005设计为穿过血脑屏障,以将治疗浓度的紫杉醇递送至肿瘤部位。
一项针对患有脑转移的乳腺癌患者的2期研究显示,在亚人群中,患者的中位生存期为8个月,相比之下,采用其他形式疗法的标准治疗为4个月,而未经治疗为2个月。ANG1005的其他2期研究已经完成,包括复发性高级别神经胶质瘤 ,非小细胞肺癌和脑转移瘤。目前,ANG1005已成功注册为治疗多形性胶质母细胞瘤的孤儿药物,据报道, ANG1005对抗乳腺癌的软脑膜病的3期临床试验处于募集期。
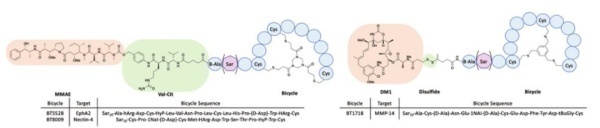
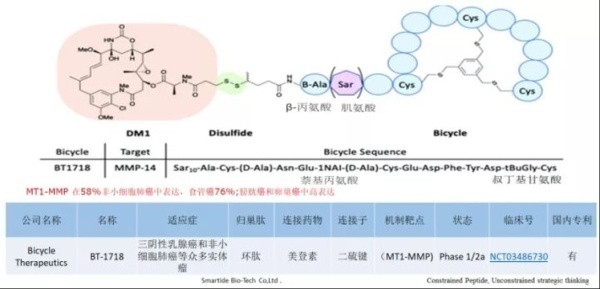
3. BT1718,BT8009,BT5528
BT1718是一种针对MT1-MMP的临床试验中的PDC,目前由Cancer Research UK赞助,处于I / IIa期。该靶点在几种肿瘤(例如卵巢癌,肺癌,乳腺癌,膀胱癌和子宫内膜癌)中过表达。BT1718由特定的双环肽组成,该双环肽过可裂解的二硫键与DM1细胞毒素偶联。I期剂量实验耐受性良好,目前进展至II期。

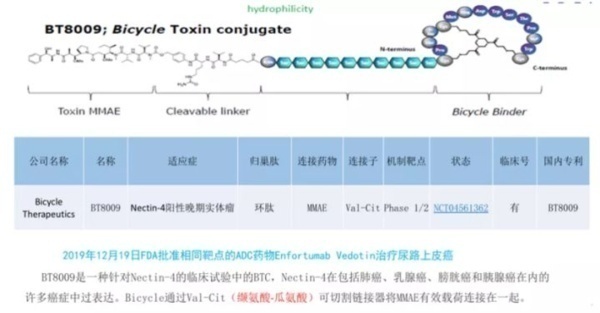
BT8009是另一种针对Nectin-4的临床试验中的PDC,Nectin-4在许多癌症(包括肺癌,乳腺癌,膀胱癌和胰腺癌) 中过表达。该PDC结构类似于BT5528,具有通过Val-Cit可裂解连接子结合在一起的MMAE有效载荷。BT8009在临床试验中显示出令人鼓舞的结果,并证明在临床前模型中具有选择性。毒性研究显示有限的脱靶毒性。
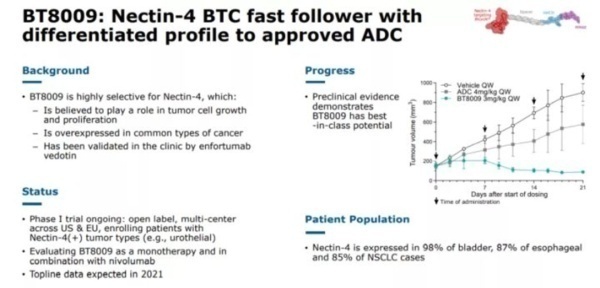
BT5528目前处理I / II期临床试验中,该药物靶向Aphrin A型受体2(EphA2),EphA2在许多肿瘤中过表达。该受体家族负责许多重要作用,包括细胞迁移,粘附,增殖和分化。BT5528由靶向双环肽组成,该双环肽通过酶可裂解的Val-Cit连接子与MMAE有效负载结合。I / II期临床试验中显示出剂量耐受良好。
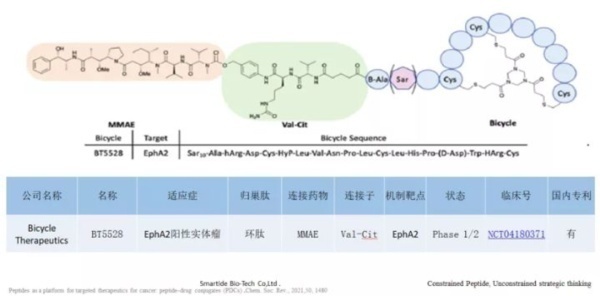

4. TH1902,TH1904
Theratechnologies开发的新型肽可与成熟的抗癌药物(如多西他赛,阿霉素或酪氨酸激酶抑制剂)结合,以特异性靶向Sortilin受体,这种方法具有改善药物安全性和有效性的潜力。该技术是2019年2月Theratechnologies以$5.3Mn收购 Katana Biopharma(2016年成立)获得的,与此同时还获得了后者的PDC产品管线,包括TH1902(多西他赛-多肽偶联物)和TH1904(阿霉素-多肽偶联物),TH1902临床前数据显示其在多种肿瘤中具有良好的疗效,且无系统毒性。
5. EP-100
EP-100是由Esperance Pharmaceuticals 公司开发的一种合成的裂解肽,靶向癌细胞上的促性腺激素释放激素 (gonadotropin-releasing hormone, GnRH)受体。这种合成肽由GnRH 天然配体与带有18 个氨基酸的阳离子α- 螺旋裂解肽连接而成。阳离子肽与带负电荷的肿瘤细胞膜相互作用,并且在几分钟内裂解细胞膜使细胞死亡。目标适应症包括乳腺癌、前列腺癌、卵巢癌、子宫内膜癌、胰腺癌、肝癌、皮肤癌、血液癌(白血病和淋巴瘤)和睾丸癌。
在原位癌小鼠模型中,EP-100 促使卵巢癌细胞数量显著下降,减缓移植肿瘤模型中的肿瘤生长速度,并解决了L- 氨基酸肽在体内降解的问题,为实体肿瘤的治疗提供了安全可行的替代方案。
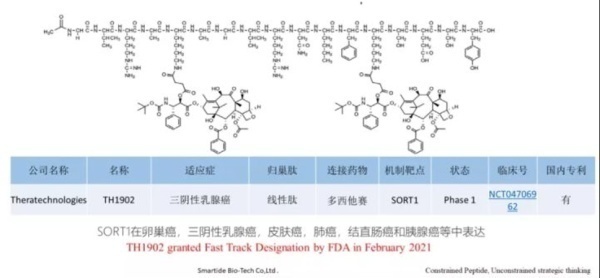
6. SBI-1301
SBI-1301是Soricimed Biopharma公司开发的一款PDC药物,其正在利用其对TRPV6靶向肽的开发用于癌症治疗的下一代肽-药物偶联物。用于癌症治疗的有效的杀癌药物无法将肿瘤组织与健康组织区分开来,从而导致狭窄的治疗范围和严重的毒性。为了克服这些局限性,公司将这些高效的细胞毒性有效载荷与靶向TRPV6的肽连接起来,并将有效载荷快速而直接地传递至肿瘤。
7. CBP-1008
CBP-1008和CBP-1018即采用容易内吞的叶酸受体(FR),肿瘤特异性靶点则分别选用TRPV6和PSMA。从同宜医药公布的专利来看,其整体开发思路是利用叶酸作为配体,再利用另外已报到的天然肽作为第二配体进行规避专利。例如TRPV6配体即是利用Soricimed 公司开发的SOR-C27中的SOR-C13,SOR-C13也是Soricimed公司开发的一款活性多肽,目前处于临床研究阶段。同宜医药使用的连接子和有效载荷与BT8009与BT5528一致。同宜医药目前在自主研发PDC药物处于国内前列,其后续临床进展和国际化进程,值得持续关注。
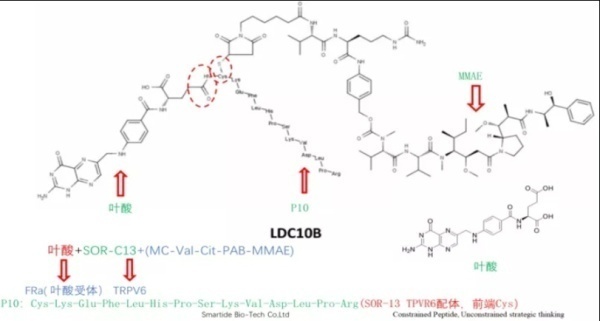
8. BGC0228
2021年12月7日,CDE官网显示,博瑞医药BGC0228临床试验申请首次获药监局批准,用于治疗晚期恶性实体瘤。BGC0228是博瑞医药开发的PDC药物,拟开发用于肿瘤治疗。BGC0228是由疗效明确的拓扑酶I抑制剂与具有肿瘤靶向的多肽结构偶联而成:BGC0228能靶向肿瘤组织高度表达的CD44,使药物在肿瘤部位富集,临床拟开发用于小细胞肺癌,胰腺癌,结直肠癌,乳腺癌等多种实体瘤的治疗。
05 PDC药物公司
代表性企业
1. 日本PeptiDream及其PDPS平台
PeptiDream是一家位于日本的生物制药公司,成立于2006 年,致力于解决全球患者未被满足的医疗需求,并提高他们的生活质量。该公司专有的多肽发现平台系统(PDPS)能以高效率生产高度多样化的非标准多肽库,用以鉴定高效和选择性命中候选物,并开发成基于多肽、小分子或多肽-药物偶联物(PDC)的疗法。目前,PeptiDream已经与诺华、拜尔等多家国际制药企业就PDC药物研发签订合作协议。
PeptiDream核心技术为灵活地合成“非标准肽”(Flexizyme 技术),在一个小管(FIT 系统)中生成数万亿种类型的综合化合物库,以高速筛选苗头药物。这些技术结合起来构成了公司的专有的肽发现平台系统(PDPS),供制药企业使用。
Peptidream 公司独有的多肽发现发现平台系统:PDPS(Peptide Discovery Platform System)是面向下一代创新 药物的发现平台,它基于三项关键技术:
1) Flexizyme 技术和PDTS(肽发现翻译系统)
传统核糖体合成多肽主要是通过tRNA的转运作用和tRNA与核糖体间碱基对互补作用将氨基酸依次连接到肽链上的,这种过程通常发生在生物体细胞内,这个过程包括两个重要反应:1. 氨酰-tRNA合成酶(ARSs)催化氨基酸连接到各自的tRNA上;2. tRNA与mRNA在核糖体识别配对。其中第一步反应可以应用于体外合成多肽:采用重组ARSs可将特殊氨基酸(AAs)与tRNA 结合,但底物杂交率低下。
而Flexizyme能高效催化天然氨基酸和特殊氨基酸与tRNA结合,合成tRNA-氨基酸复合物,以备下一步进行翻译。PDTS(Peptide Discovery Translation System,肽发现翻译系统)是上述第2 个关键反应的合成系统:通过核糖体合成肽的无细胞转录/翻译系统。当PDTS被单独使用时,它只能合成由20种天然氨基酸组成的肽,但当它与Flexizyme技术相结合时,可以使用400多种特殊氨基酸合成各种肽。
2) 肽环化技术和改性技术
PDPS 的另一项重要构建技术是:环化各种链肽并将其转化为特殊环肽。肽环化技术通过改变肽链一端的基团,使其能通过化学反应收尾相连形成环肽。重要步骤为
a. 用N-2-氯乙酰氨基酸取代N-端(起始密码子);
b. 分子内硫醚与下游半胱氨酸键的形成;
c. 自发环化(甚至可能发生在核糖体内)。特殊环肽与常见线性肽相比,具有很多优点,如结构更加刚性、对靶蛋白有更高亲和力和选择性、在体内更稳定等。
3) PD显示(PeptiDream Display)
上述两种技术使得快速制备多种肽库成为可能,接下来就需要从包含数千种肽的库中选择可能有治疗作用的命中肽。与其他显示技术相比,PD显示能够在短时间内识别筛选出命中肽,并具有极高的重现性和重复性。公司对PD显示不断进行优化,以配合正在研究的肽流技术。
2. Soricimed
Soricimed正在利用其对TRPV6靶向肽的知识来开发用于癌症治疗的下一代肽-药物偶联物。用于癌症治疗的有效的杀癌药物无法将肿瘤组织与健康组织区分开来,从而导致狭窄的治疗范围和严重的毒性。为了克服这些局限性,公司将这些高效的细胞毒性有效载荷与靶向TRPV6的肽连接起来,并将有效载荷快速而直接地传递至肿瘤。结果,该抗癌药迅速在肿瘤中积聚,并使健康组织免于暴露于这些有毒物质。
与其他药物偶联程序(例如抗体-药物偶联物(ADC))相比,肽-药物偶联物(PDC)方法具有多个优势。与ADC相 比,公司的PDC更小,具有更好的肿瘤渗透性,更低的全身暴露,更低的肝损伤风险,并且制造更容易,更便宜。SBI-1301是Soricimed最先进的PDC之一,它是一种极强的细胞毒性剂的结合物,在小鼠异种移植的前列腺癌中,仅用12天的3种治疗就已显示出完全的肿瘤消退。在测试的最高剂量下,没有明显的毒性症状。观察小鼠60天,并且肿瘤没有重新生长。
06 其他偶联药物
SMDC
小分子偶联药物(Small molecule-drug conjugates,SMDC)是由小分子的靶向配体与细胞毒药物偶联所得。与ADC结构部分类似,构成的关键因素有三点:小分子靶向配体、细胞毒分子和连接子。其作用机制也与ADC类似,但SMDC能更快速均匀地分散到肿瘤组织中,且成本低、无免疫原性。
目前SMDC仍未有上市药物,其研发难点主要是小分子配体难以获得,限制了其发展。2014年,默沙东公司与Endocyte公司合作的SMDC药品vintafolide(长春碱-叶酸偶联物)曾获得有条件上市,但是其III期临床试验失败(并未公布具体原因),默沙东与Endocyte从欧盟撤回vintafolide的有条件上市许可申请。
直至今日,SMDC药物研发的历史并不算长,国内外也只有少数几家公司在该领域有所布局,国外代表性公司有Endocyte、Tarveda、Bind Therapeutics,国内仅博瑞生物有公开信息显示布局SMDC药物研发。
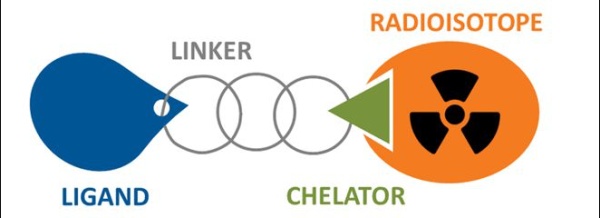
RDC
放射性核素偶联药物(Radionuclide Drug Conjugates,RDC)主要由靶向配体、连接子、螯合物和细胞毒/成像因子(放射性同位素)组成。
RDC与ADC和SMDC的最大差异在于药物载荷。RDC荷载的不再是小分子,而是放射性核素,使用不同的医用核素,可以具备显像或治疗等不同功能,部分核素兼备两种功能。基于这些功能,RDC将有可能成为肿瘤诊断、成像以及治疗的新技术。
目前,国外布局该领域的由诺华、拜耳、加尼福尼亚大学、RadioMedix和Curium等企业或机构;国内的企业,则有远大医药在这一赛道有较多的布局。
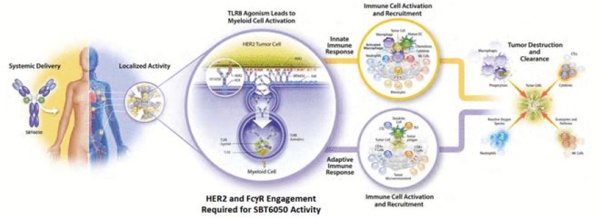
ISAC
2021年8月,信达生物与Bolt(纳斯达克股票代码: BOLT)达成合作,宣布引入Bolt公司的抗体-免疫刺激偶联物( Immune-stimulating Antibody Conjugate ,ISAC)技术,共同开发三款候选药物。ISAC是一类在功能上与ADC部分类似的偶联药物,它的不同之处在于可以驱动先天免疫和特异性免疫,还可通过TLR激活CD4/CD8 T细胞将冷肿瘤转化为免疫热肿瘤。ISAC通过调节肿瘤的免疫微环境和免疫刺激,达到治疗肿瘤疾病的目的。
目前,国外布局该领域的研发公司有诺华、Bolt Biotherapeutics、Silverback Therapeutics、Mersana Therapeutics;国内除了信达生物通过合作进行研发外,恒瑞医药和百济神州通过自主研发也在积极布局该领域。
ADeC
瑞士公司Debiopharm和韩国Ubix Therapeutics达成合作研究,将结合两家专有的技术平台MultilinkTM和Degraducer®开发抗体降解偶联药物(Antibody Degraducer Conjugates ,ADeC)。将要开发的ADeC药物会是一款将载荷替换为降解分子的抗体偶联药物,或许也会同时携带其他载荷实现协同作用。ADeC目的也是将降解分子携带至靶位置,避免全身暴露,甚至克服一些Protac分子潜在成药性问题,如理化缺陷、 特异性、PK等。
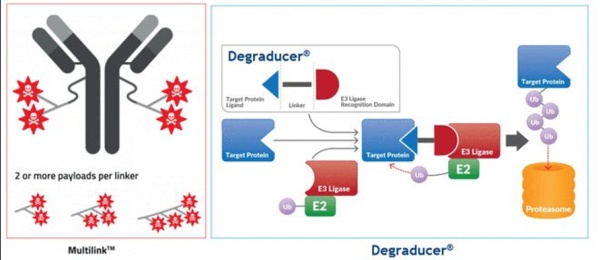
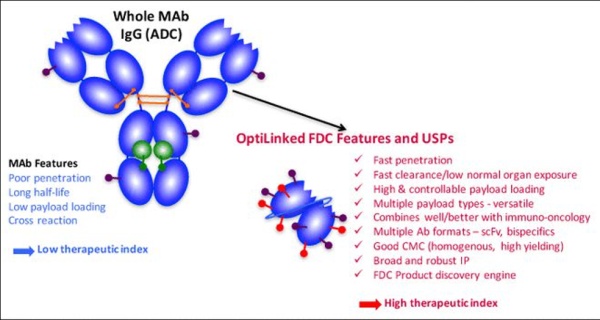
FDC
抗体片段偶联药物(Antibody fragment-drug conjugates ,FDC),顾名思义,就是使用更小的抗体片段(单链 scFv)替换更大的抗体分子。通常认为,抗体片段相对容易发现,并且可以采用生物工程技术,实现更高的DAR。FDC与ADC技术上几乎相通,但采用更小的片段抗体,有望提高肿瘤渗透,最大化药物效力。小片段和缺乏Fc能够在正常组织和循环中快速清除,降低毒性。
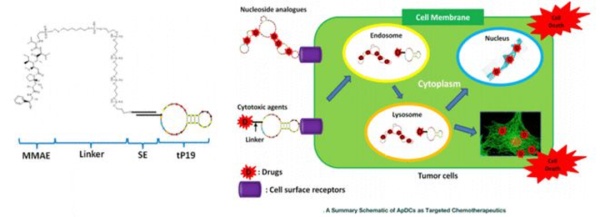
ApDC
适体偶联药物(aptamer drug conjugates,ApDC)是一种采用结构化的寡核苷酸序列作为靶向相应分子的偶联药物形式。核酸适配体被称为“化学抗体”,具有与抗体相似的靶向性和靶标结合力。相较于抗体,核酸适配体也具有诸多优点,比如稳定性高、免疫原性小、生产成本低、化学修饰容易等。由于ApDC药物采用寡核苷酸序列,在Linker链接和偶联策略上或许和ADC药物存在差异,但在药物组成以及作用机制和载荷与ADC药物并无太多区别。
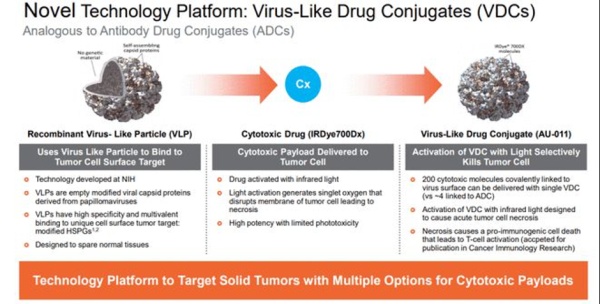
VDC
病毒样药物偶联物(virus-like drug conjugates,VDC)是采用将病毒衣壳设计为非感染性蛋白质纳米颗粒(病毒样 颗粒,VLP)作为高效递送载体的偶联药物形式。
Aura采用源自人乳头瘤病毒或HPV的VLP选择性地附着在修饰的硫酸乙酰肝素蛋白聚糖(HSPG)表面,实现与实体肿瘤细胞或转移灶结合,而不与正常组织结合的目的。AU-001正是这种机制的VDC产品,病毒样成分选择性地结合 HSPG,偶联的红外光激活细胞毒药物被激活后,选择性破坏肿瘤细胞,导致肿瘤细胞急性坏死的同时激活免疫系统产生抗肿瘤应答。
AOC
抗体寡核苷酸偶联物(Antibody-oligonucleotide conjugates,AOC)是指利用抗体将治疗性寡核苷酸(siRNA 、PMO等)递送至特定细胞或组织,从而减少治疗患者疾病所需的药物量以及解决不可靶向和寡核苷酸递送问题。寡核苷酸与靶向配体的偶联还可以改善寡核苷酸(治疗性RNA或DNA分子)的药代动力学特性并扩大其应用范围。与适体偶联物(ApDC)相反,AOC的目的是实现寡核苷酸的靶向递送,阿斯利康曾开展过相关产品的研究。
从技术上说,AOC采用的是抗体作为递送介质,也可以认为小分子(包括多肽)、蛋白质(酶)等也能实现相关功能。如果细分,仅以寡核苷酸为载荷的药物也会衍生出多种概念性产品。
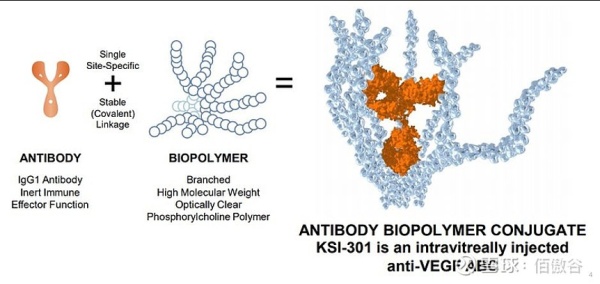
ABC
抗体-生物聚合物偶联物(ABC)
AMD(年龄相关性黄斑变性)为眼科领域的下一个蓝海,拥有庞大的患病群体。而当下抗VEGF药物在眼科市场占据相当大的比重,已然成为其主力军,但现有的抗VEGF药物通常需要通过玻璃体内给药,且在玻璃体内半衰期短, 因此患者的依从性低下。Kodiak公司基于专有的ABC技术平台开发的一种新型抗VEGF生物制剂-KSI-301,该公司通过将生物聚合物与抗体偶联,提高药物在眼睛内的持续时间,提高疗效并减少注射频率。
AAC
抗体-抗生素偶联物( AAC)
目前大多数抗生素对细胞外病原菌存在的杀伤效率低下,有研究者提出专门针对细胞内病原菌的治疗策略来促进治疗效果。早在2015年,Lehar的团队首次提出和评估了抗体-抗生素偶联物(AAC),该研究团队设计出一种anti-β-GlcNAC WTA(金黄色葡萄球菌表面的糖类分子)抗体—利福平类抗生素的AAC,其抗体部分可与游离的金黄色葡萄球菌结合,然后通过Fc介导的调理吞噬作用进入吞噬细胞内部,而后利福平类抗生素被释放出来并将隐藏在吞噬细胞内的金葡菌杀死。在小鼠感染模型中,在感染后24h分别给予AAC药物和万古霉素治疗后,研究结果表明,与万古霉素相比,AAC药物能够更好地清除体内的金黄色葡萄球菌。
来源:金佳路 倚锋资本

来源:网络
版权及免责声明:本网站所有文章除标明原创外,均来自网络。登载本文的目的为传播行业信息,内容仅供参考,如有侵权请联系答魔删除。文章版权归原作者及原出处所有。本网拥有对此声明的最终解释权。
{replyUser1} 回复 {replyUser2}:{content}